PART 1. МЕТАБОЛИЗМ ЖЕЛЕЗА. Многое забыто, а что-то даже на разбирается в школьной (университетской) программе.
Железо является основным элементом, вовлеченным в различные жизненные функции: транспорт кислорода, синтез дезоксирибонуклеиновых кислот, метаболизм энергии и клеточное дыхание. Несмотря на это, избыток железа в организме приводит к окислительному стрессу, окислению липидов и дезоксирибонуклеиновых кислот, что в конечном счете приводит к клеточной смерти.
Гомеостаз железа в организме поддерживается за счёт взаимодействия нескольких систем:
-эритроциты и пролиферативные клетки. Содержат 2-3 г железа.
-энтероциты (в которых железо всасывается) и селезеночные макрофаги (которые поглощают гемовое железо из разрушенных эритроцитов)
-гепатоциты (которые являются хранилищем). Содержат 1 г железа.
-плазма крови содержит всего лишь 3-4 мг железа.
У женщин запасы железа часто могут быть истощены (дефицит железа), вследствие плохого питания и обильной менструации.
Почти всё железо путешествует по организму с помощью трансферрина (Tf). Трансферрин может существовать в трёх формах:
-апотрансферрин(Apo-Tfr) без атомов железа.
-монотрансферрин с одним атомом железа.
-дитрасферрин с двумя атомами железа.
Максимально одна молекула трансферрина может переносить два атома железа. Подходя к клеткам, которые нуждаются в железе, трансферрин связывается с рецептором- рецептор трансферрина 1(TfR1) и погружается внутрь клетки вместе с рецептором в составе эндосомы. Для того, чтобы атомы железа могли освободиться от связи с трансферрином, внутрь эндосомы с помощью протонной помпы загоняются протоны водорода и создаётся кислая среда, благодаря чему атомы железа освобождаются от трансферрина и дальше могут выйти из эндосомы в цитоплазму клетки. Эндосома вместе с рецептором и трансферрином встраивается обратно в клеточную мембрану. Пустой трансферрин идёт дальше по своим делам, а рецептор готов ловить следующий «полный» трансферрин.
Ниже представлена схема. Смотреть слева направо)
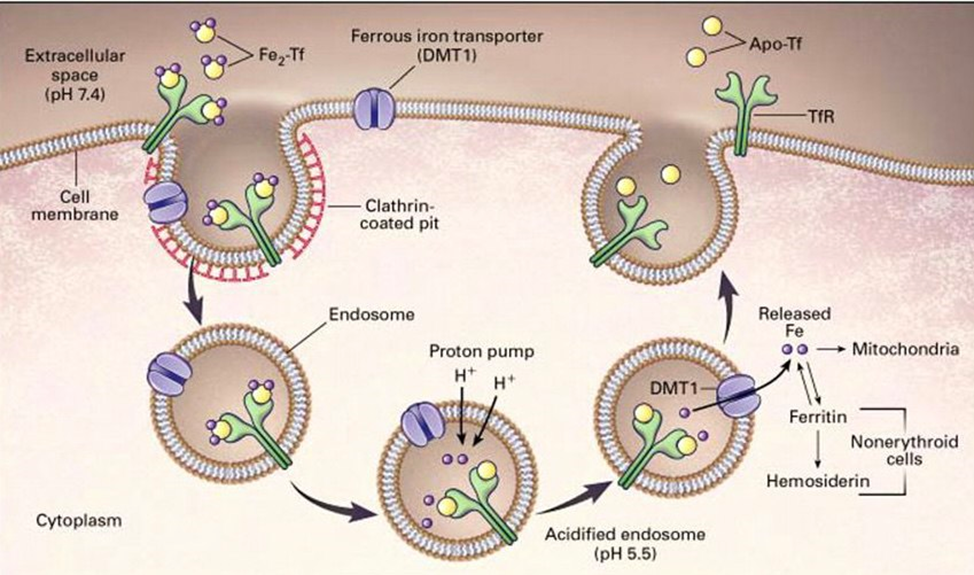
Поскольку у человека в организме не существует механизмов для выведения железа, природа отыгралась на создании крайне сложного, многофакторного механизма регуляции поступления железа.
В основном, баланс железа контролируется уровнем абсорбции его в проксимальной части двенадцатиперстной кишки. Поглощение железа энтероцитами увеличивается, когда усиливается эритропоэз и снижается, когда запасы железа в организме переполнены. Только 1-2 мг железа поступает через кишечник, что эквивалентно дневной потере железа, в основном, постоянный уровень необходимого железа поддерживается за счёт его повторного использования с помощью поглощения макрофагами «старых» эритроцитов (20-25 мг в день).
Вернёмся к тому, что происходит с железом в кишечнике. Вместе с пищей железо поступает в организм со степень окисления Fe+3 , поскольку такое железо крайне малодоступно для всасывания, оно восстанавливается с помощью мембран-ассоциированной железоредуктазы до Fe+2 и впоследствии транспортируется через апикальную мембрану двухвалентным металлическим транспортером (divalent metal transporter DMT-1 ). DMT-1 основной трансмембранный транспортер железа и его экспрессия сильно индуцируется при железодефицитных состояниях. Бо‘льшая часть железа попадает в организм вместе с гемом с помощью белка переносчика гема 1 (HCP1). Затем гем катаболизируется микросомальной heme oxygenase 1 (HO-1) в биливердин, высвобождая Fe2+. Гемовое железо обладает большей биодоступностью и является важным источником железа в организме, но механизм обмена гема до конца не изучен.
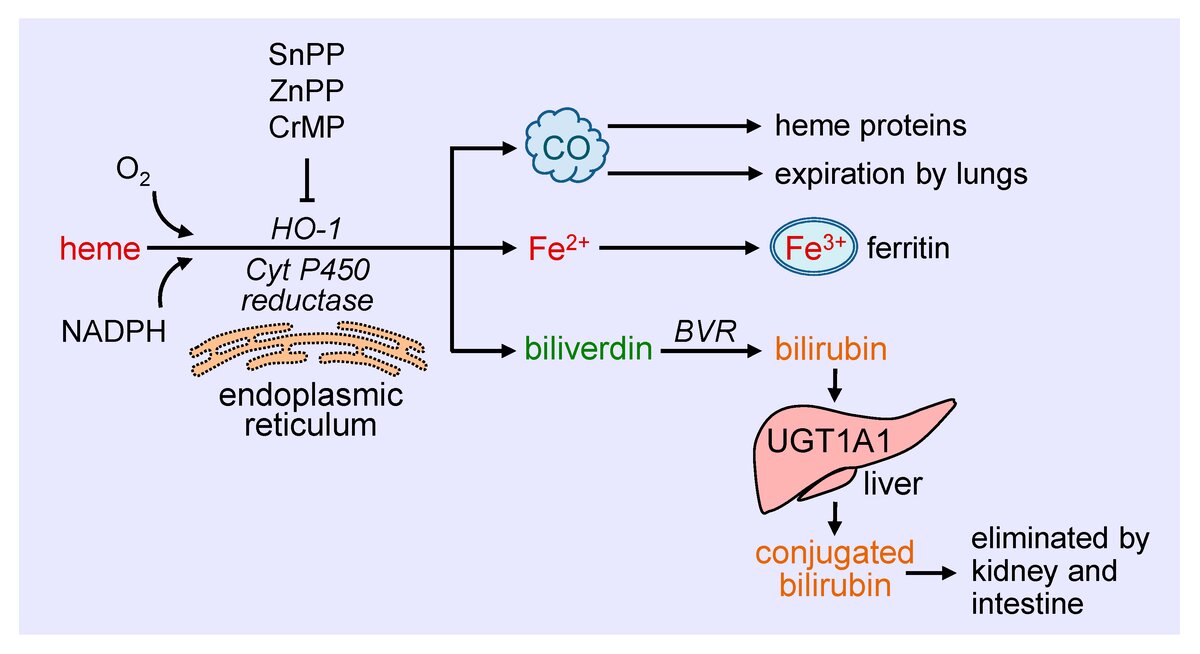
Часть железа остаётся внутри клетки на хранение, другая часть переносится через базолатеральную мембрану энтероцита с помощью мембранного транспортера ферропортина(FPN). Через ферропортин железо выходит в степени окисления Fe+2, которое активно может проявлять свои окислительно-восстановительные свойства и приводить к повреждению клеток, поэтому после выхода из энтероцита, сразу же на мембране, железо окисляется с помощью белка гефестина (Hp) до степени окисления Fe+3.
Абсорбция железа в двенадцатиперстной кишке регулируется двумя путями: автономно клеточным и системным. Автономный механизм основан на взаимодействии белков, регулирующих обмен железа iron regulatory proteins (IRPs), которые после транскрипции регулируют экспрессию основных белков метаболизма железа и гипоксия чувствительной механизм, основанный на транскрипционных факторов, которые индуцируются гипоксией , в частности HIF-2a. В условиях повышенной потребности железа HIF-2a может повышать экспрессию мембран-ассоциированной железоредуктазы , DMT1 и ферропорина (FPN).
На системном уровне, взаимоотношение IRP-1/HIF-2a координирует всасывание железа в кишечнике в зависимости от системной потребности в кислороде и железе.
Железо, высвобождаемое из энтероцитов и макрофагов захватывается циркулирующим апотрансферрином и становится монотранферинном, а когда железо в крови в избытке, монотрансферрин захватывает второй атом железа и становится дитрансферрином. Далее трансферрин связывается с рецептором трансферрина 1(Tf1) по выше описанному механизму (см рис 1).
Насыщение железом трансферрином плазмы крови (Iron saturation of serum transferrin TSAT) является главным индикатором, определяющим системный гомеостаз железа. TSAT определяется количеством железа, абсорбированным в кишечнике, рециркулируемого и высвобождаемого из макрофагов и используются для эритропоэза- основного потребителя железа. Насыщение трансферрина определяется в процентах и в норме равно 15-30%. Также, комплекс железо-трансферрин, влияет на экспрессию гормона гепсидина, который регулирует абсорбцию железа в кишечнике и высвобождение из макрофагов через посттранскрипционную регуляцию экспортера железа ферропортина (FPN). Когда красный костный мозг перенасыщается железом, уровень гормона гепсидина повышается, он воздействует на белок ферропортин(FPN) и блокирует выход железа из энтероцитов и макрофагов. В следствие чего уровень железа плазмы крови снижается. Железо и воспаление являются основными индукторами гепсидина. Гепсидин - один из белков острой фазы воспаления и является одним из важных механизмов борьбы с бактериально и грибковой инфекцией, т.к бактерии и грибки( в частности Escherichia coli, Neisseria cinerea, Staphylococcus epidermidis, Staphylococcus aureus, Streptococcus agalactiae, Candida albicans) для процессов своей жизнедеятельности тоже используют Fe3+, соответственно: чем ниже уровень железа в кровотоке, тем меньше шансов у бактерий размножиться. Условно говоря, гепсидин запирает железо в макрофагах и энтероцитах, чтобы бактерии и грибки не могли его получить, что приводит к снижению уровню железа в анализе крови. При повышении запасов железа, уровень гепсидин возрастает благодаря активации bone morphogenetic proteins(BMP6), который после запуска каскада реакций SMAD- тормозит всасывание железа в кишечнике.
Экспрессия гепсидина подавляется при железодефиците, гипоксии и при повышенной работе красного костного мозга. Ингибиторами гепсидина являются: matriptase 2, кодируемый геном transmembrane serine protease 6 (TMPRSS6), эритропоэтин и эритроферон (ERFE).
При дефиците железа matriptase 2 ингибирует гепсидин путем расщепления ко-рецептора BMP6 гемоювелина(HJV) на мембранах гепатоцитов. Генетические мутации в гене TMPRSS6 приводят к неконтролируемому повышенному уровню гепсидина и, как следствие, врожденной микроцитарной анемии. Эритроферон(ERFE) индуцируется эритропоэтином(EPO), что приводит к снижения уровня гепсидина и опосредованному повышению уровня железа крови.
Ген TMPRSS6 играет ключевую роль в определении уровня гепсидина.
(Чтобы понять механизм, нужно посмотреть все эти схемы несколько раз)
Внутриклеточное железо, если не используется на всякие разные нужды, запасается в ферритине или экспортируется из клетки с помощью ферропортина (если его слишком много) чтобы избежать токсического эффекта.
Ферритин -это хранилище для железа, которое сберегает его для разных клеточных нужд, а также предотвращает развитие окислительного стресса. Одна молекула ферритина может содержать до 4500 атомов Fe. В клинической практике мы видим, что уровень ферритина снижается при железодефицитных состояниях и повышается при воспалении, т.к атомы железа продолжается поступать в клетку, но заперты в ней с помощью ферропортина из-за того, что гепсидин заблокировал белок ферропортин.
Во время беременности ферропортин в большом количестве экспрессируется на базальной мембране синцитиобластов плаценты и играет важную роль в передаче делеза плоду. Плацентарный ферропортин строго регулируется концентраций железа в трофобласте через систему IRE-IRP. Интересно, что уровень плацентарного гепсидина крайне низок и почти не регулирует обмен железа матери и плода. Несмотря на это, уровень системного гепсидина при инфекции и воспалении будет повышаться, через систему интерлейкина-6, и приведет к общему дефициту железа у матери и плода.
Мутации, приводящие к потере положительной регуляции гепсидина, приводят к увеличение всасывания железа в кишечнике, излишнему накоплению его в запасах и образование свободного железа в плазме крови несвязанного с трансферрином.
Мутации, приводящие к потере отрицательной регуляции гепсидина (мутации в гене TMPRSS6 ), приводят к стойкому повышению уровня гепсидина и, как следствие, к развитию железорефрактерной железодефицитной анемии(IRIDA).
Дефицит железа, железодефицитная анемия и анемия хронического воспаления часто встречаемые заболевания в популяции. Дефицит железа может приводить к нарушению эритропоэза до развития железодефицитной анемии. Железодефицитный эритропоэз часто встречается у женщин в пременопаузе, у которых по факту уровень гепсидина тоже снижен.
Дефицит железа или нарушение всасывания железа и утилизации в костном мозге вызывают железодефицитную анемию.
Анемия воспаления, которая является второй по частоте анемией в мире, характеризуется секвестрацией железа и снижением доступного железа для эритропоэза.
Высокий уровень гепсидина ассоциирован с новообразованиями, хроническими инфекциями и хроническими заболеваниями почек и приводи к анемии хронического воспаления.