
Вышло очень хорошее ревью на тему аутоиммунной симптоматики при первичных иммунных дефицитах. Часто это причина ложного ревматологического диагноза, вместо верного иммунологического.
первичные иммунные дефициты многогранны в своих проявлениях. На сегодня нам известно уже около 600 мутаций в генах иммунной системы, что приводят к первичным иммунным дефицитам разной тяжести. Помимо наличия мутации, есть еще фактор пенетрантности, то есть мутация может быть, но может быть компенсирована каким-то механизмом (моноаллельная экспрессия, например), но клинические проявления будут стерты или атипичны.
Тут авторы проходят через все типы ПИДов, комментируя долю ревматологических-аутоиммунно-воспалительных симптомов, что могут скрывать за собой иммунные дефициты. Среди них васкулиты, скелетно-мышечные симптомы, температура, сыпи, аутоиммунные цитопении. Помимо ревмо симптомов, тут нерпоходящие энтериты, настриты, гепатомегалия и аутоиммунный гепатит, а значит гастроэнтерологи, различные дерматологические нарушения, эндокринные, почечные, легочные и тд.
Авторы приводят говорящие примеры:
Пациенты с аутовоспалительным синдромом, ассоциированным с NLRC4, которые обращаются с болями в суставах, лихорадкой и сыпью, могут быть обследованы ревматологами на предмет системной красной волчанки (СКВ) или системного ювенильного артрита.
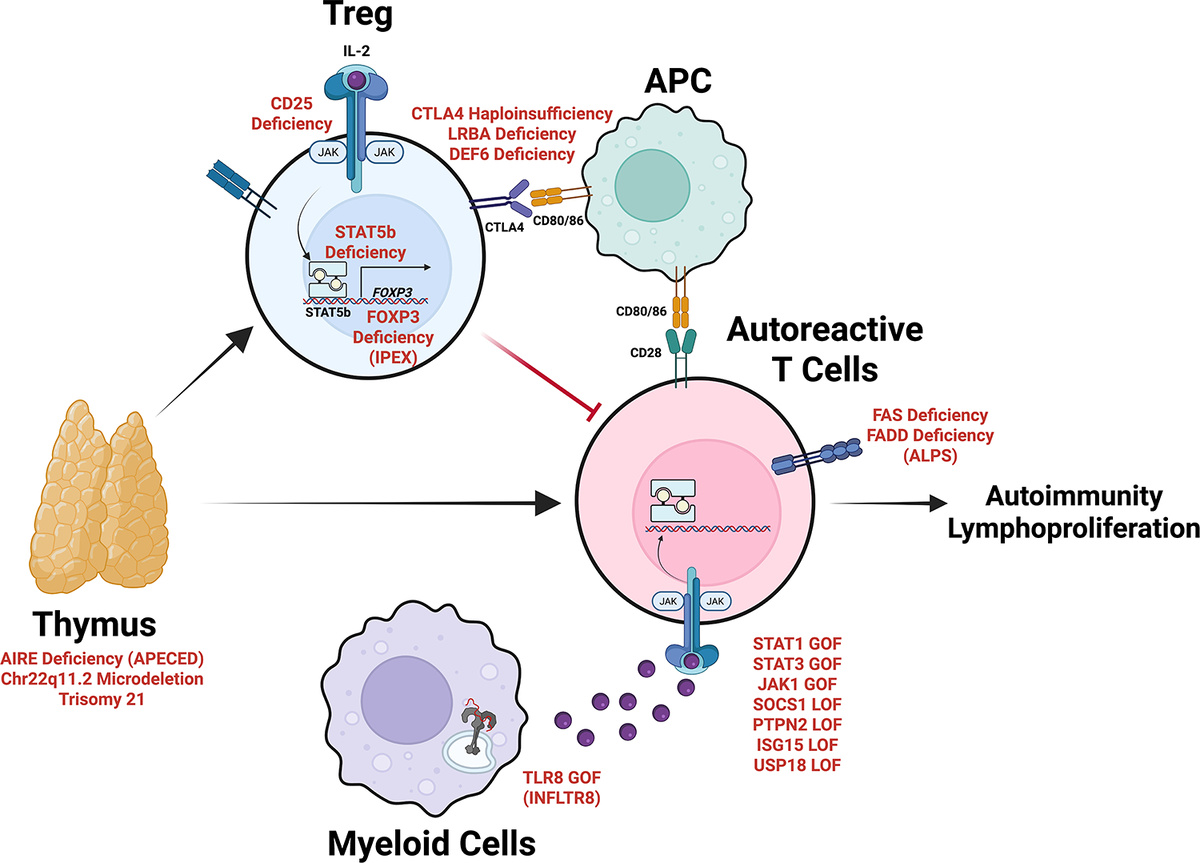
Пациенты с гаплонедостаточностью CTLA4 могут обратиться к ревматологу с болями в суставах и интерстициальным заболеванием легких, но также иметь значительный дефицит антител, который важно распознать.
ПИДы с преимущественной восприимчивостью к инфекциям
1. Тяжелый комбинированный иммунодефицит (ТКИД):
- Определяется дефект производства иммунных клеток.
- У 25% пациентов наблюдается "негерметичный" ТКИД из-за гипоморфных вариантов (когда остаточная функция гена имеется, проявления атипичные, часто с атоиммунных нарушений)
- Может проявляться синдромом Оменна с увеличенными лимфоузлами, гепатоспленомегалией, повышенным IgE и эритродермией
2. Комбинированные иммунодефициты (КИД):
- Hyper-IgM syndrome (CD40L, CD40, AICDA, UNG, MSH6, CTNNBL1, APRIL) –повышенный риск диабета, аутоиммунные цитопении (тромбоцитопению, аутоиммунную гемолитическую анемию).
- КИД с гранулемой/аутоиммунностью может проявляться васкулитом, аутоиммунными цитопениями, миастенией
- Активированный синдром PI3K дельта (АПДС) – Часто имеет фенотип, напоминающий общий вариабельный иммунодефицит, более 30% пациентов имеют аутоиммунность, чаще всего аутоиммунные цитопении
- Расстройства активации Т лимфоцитов (ZAP70, SLP76, LAT) – воспалительные артриты, воспалительные заболевания кишки.
- Синдром микроделеции Chr22q11.2 – повышенная вероятность ювенильного артрита, аутоиммунной тромбоцитопении.
- Синдром Вискотта-Олдрича (WASP) – васкулиты, гломерулонефриты, аутоимунная гемолитическая анемия
3. Пиды с дефицитом антител (гуморальное звено)
- ОВИН (общий варьябельный иммунный дефицит) (TNFRSF13B (TACI), TNFRSF13C (BAFFR), CD19, CD21, CD81, NFKB1, NFKB2, PTEN)– СКВ, ВЗК, хроническое заболевание легких, заболевание печени (гепатит C, гранулемы, идиопатическое заболевание печени). Около 68% пациентов ОВИН после 40 лет иммеют хотя бы одно аутоиммунное проявление.
- Агаммаглобулинемия (BTK, SPI1) – ВЗК, ревматоидный артрит, усталость, хроническая диарея, сыпь, боль в суставах.
ПИДы с преимущественными ревматологическими проявлениями
1. Расстройства иммунной регуляции:
- Аутоиммунная полиэндокринопатия-кандидоз-эктодермальная дистрофия (АПЭКЭД) – мутации в AIRE, первоначально описана как триада: хронический кожно-слизистый кандидоз, гипопаратиреоз, первичная надпочечниковая недостаточность, может включать ЮИА
- Аутоиммунный лимфопролиферативный синдром (АЛПС) – обусловлен дефектами в генах FAS или FADD, пациенты имеют лимфопролиферацию и аутоиммунные цитопении
- Синдром IPEX – вызван мутациями в FOXP3, поражает преимущественно мужчин, характеризуется аутоиммунной энтеропатией, диабетом 1 типа, дерматологическими проявлениями. Экзема, дерматит, алопеция, псориазоподобные поражения, мультиорганная лимфоцитарная инфильтрация, гипопаратиреоз, ЮИА
- Расстройства цитокиновой сигнализации (STAT1, STAT3, STAT4, STAT6, JAK1, SOCS1, PTPN2, ISG15, USP18) – мутации в генах STAT могут вызывать различные аутоиммунные проявления: STAT1 GOF синдром связан с высокой частотой аутоиммунности (примерно 37% пациентов). STAT3 GOF синдром часто проявляется лимфопролиферативным заболеванием и цитопениями
- Гемофагоцитарный лимфогистиоцитоз (PRF1, STX11, STXBP2, UNC13D) – лизорадка, цитопении, повышенный ферритин, гипертриглицеридемия, энцефалит.
2. Врожденные иммунные дефекты
- Моногенная красная волчанка, дефициты ранних компонентов классического пути комплемента (C1q, C1r, C1s, C4) связаны с моногенной волчанкой почти у всех пациентов, гены деградации нуклеиновых кислот (DNASE1, DNASE1L3) также могут вызывать моногенную волчанку
- Дефекты белков-регуляторов комплемента (CFH (Factor H), CFI (Factor I), CD45, Autoantibodies targeting Factor H)– васкулопатии, аГУС.
3. Аутовоспалительные расстройства:
- Интерферонопатии 1 типа (TREX1, RNAASEH2A, RNASEH2B, RNASEH2C, TREX1, ADAR, IFIH1), COPA, STING1, ADA2) – Синдром Айкарди-Гутьера (АГС) и связанные состояния, пациенты часто имеют тяжелые неврологические нарушения и аутовоспалительные кожные проявления
- Инфламмасомопатии (MEFV (FMF), NLRP3, PLCG2, NLRC4) – семейная средиземноморская лихорадка (мутации MEFV), криопирин-ассоциированный периодический синдром (мутации NLRP3), характеризуются периодическими лихорадками и воспалением различных органов. Перитониты, плевриты, синовиты.
- Неинфламасомные воспалительные расстройства (TNFRSF1A (TRAPS) TNFAIP3 (A20), RELA, IKBKG)– Оральные и генитальные язвы, артрит, узловатая эритема, рецидивирующие лихорадки, ВЗК с очень ранним началом
- VEXAS синдром – вызван соматическими мутациями в UBA1, проявляется у взрослых рецидивирующими лихорадками, альвеолитом, артритом, хондритом
Диагностика
Конечно тут на первый план выходит генетическое тестирование.
Показательный случай описывают авторы:
В одном иллюстративном клиническом случае пациенту с дефицитом LRBA изначально был поставлен диагноз ЮИА. Лечение НПВС, кортикостероидами и метотрексатом было безуспешным, и у пациента развились цитопении, резистентные к ВВИГ, ритуксимабу, циклоспорину и спленэктомии. Генетическое тестирование выявило дефицит LRBA, и пациент был начат на целевую терапию абатацептом, что привело к клиническому улучшению, пока она не получила трансплантацию гемопоэтических клеток (ТГК)
Генетическое тестирование должно предлагаться пациентам независимо от семейного анамнеза, и отсутствие семейного анамнеза подобного заболевания никогда не должно исключать генетическое тестирование. – это, пожалуй, главная мысль данного ревью. Если вы подозреваете ПИД, а семейный анамнез молчит– продолжайте подозревать ПИД и делайте генетику.
Второй момент, раннее начало аутоиммунности должно вызывать высокое клиническое подозрение на ПИД:
учитывая моногенную природу ПИД, пациенты часто проявляются в очень молодом возрасте по сравнению с полигенными/сложными аутоиммунными расстройствами. Например, СКВ обычно поражает людей в возрасте от 15 до 44 лет с преобладанием женщин, тогда как моногенная СКВ проявляется как у мужчин, так и у женщин, часто до 5-летнего возраста.
И третий момент, развитие множественных состояний иммунной дисрегуляции или аутоиммунность/аутовоспаление, рефрактерные к стандартному лечению – также прямое показание сделать генетику.
Авторы прекрасно описывают сложности с интерпретацией ген тестов: Несмотря на достижения в технологиях секвенирования, приблизительно 60-70% пациентов, обследуемых на ПИД, не получают генетический диагноз. Соматический мозаицизм, который возникает, когда постзиготная мутация присутствует только в некоторых клетках, был описан при множественных ВОИ, например, FAS, TLR8 и UBA1. Современные технологии секвенирования ES и WGS могут не обеспечивать достаточную глубину секвенирования для выявления низкочастотных соматических вариантов, или вариант может отсутствовать в секвенируемой ткани.
Эпигенетические механизмы также могут влиять на пенетрантность ПИД. Недавно это было продемонстрировано в семьях, гетерозиготных по болезнетворному варианту с неполной пенетрантностью из-за аутосомного случайного моноаллельного экспрессирования, приводящего к экспрессии либо нормального (здорового), либо мутантного аллеля в иммунных клетках. Такое моноаллельное экспрессирование может затруднить выявление типа наследования и определение патогенности варианта.
В общем в ревью полно интересных деталей, описаний симптоматики, что пересекается между ревмо и иммунными патологиями, и варианты подхода к проблемам, включая ген тестирование и его аргументацию. Да, ревматология сложнейшая область, где нужно знать еще и многочисленные ПИДы. А генетические результаты возьмите себе за правило регулярно пересматривать ( хотя бы раз в 3 года, если ген диагноза так и нет).
комментарии в телеграме @immunobee1