Человечеству известно более 6000 редких заболеваний. Чем реже заболевание, тем меньше компании хотят вкладываться в его лечение, ведь рынок сбыта лекарства маленький. Заболевание считается редким, если им страдает менее 1 из 2000 человек. По оценкам, каждый 17-й человек в течение своей жизни страдает от какого-либо редкого заболевания. Очень немногие из этих заболеваний излечимы, а большинство даже не имеют эффективных методов лечения.
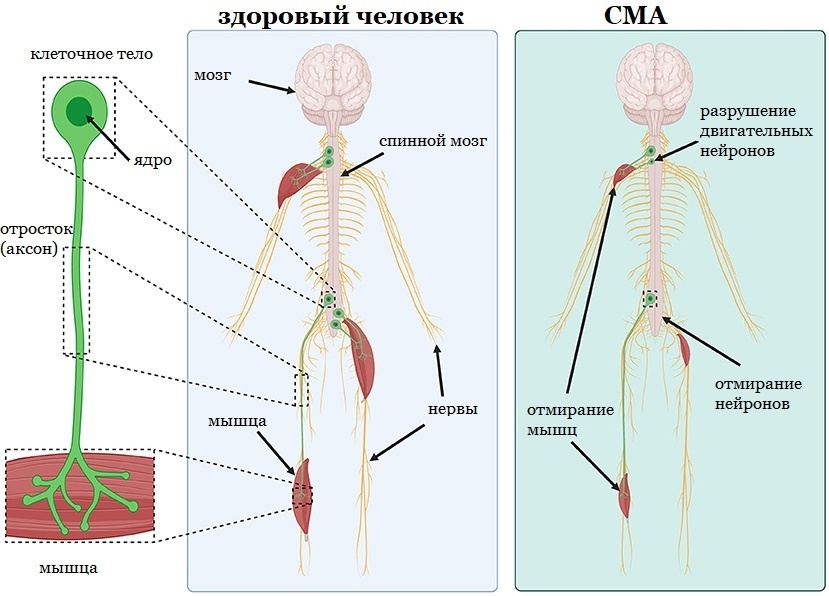
Одним из редких заболеваний является спинальная мышечная атрофия (СМА) — редкое заболевание нервной системы, которым страдает 1 из 10 000 человек. У людей со СМА наблюдается мышечная слабость и атрофия мышц, когда мышцы истощаются и усыхают из-за недостаточного использования. Первые признаки СМА обычно начинаются в возрасте до 6 месяцев и начинаются с затруднённого дыхания. Без помощи в лечении большинство детей со СМА не доживают до второго дня рождения. Всё дело в том, что двигательные нейроны не работают должным образом, начинают разрушаться и в конце концов погибают. Эти нейроны нужны, чтобы посылать электрические сигналы к мышцам, чтобы мы могли двигать телом, глотать, дышать и шевелить конечностями. Без нейронов мы не можем это делать. Двигательные нейроны состоят из командных центров, называемых клеточными телами, где находятся ядро и генетический материал клетки, и из длинного тонкого отростка, называемого аксоном, который передаёт электрический сигнал к мышце. Аксоны соединяют тело клетки с отдельными мышцами, где они образуют особые связи, которые участвуют в нервно-мышечном общении и в сокращении мышц.
СМА — генетическое заболевание, то есть у человека имеются ошибки в генах, из-за которых нейроны не работают как надо. Оно передаётся по наследству. Ошибки в генах появляются из-за мутаций, изменений последовательности ДНК под влиянием различных факторов. В случае СМА мутация находится в гене под названием "выживающий двигательный нейрон" (Survival Motor Neuron 1). Этот ген производит белок под названием SMN почти во всех клетках, но нейронам он нужен особенно. Зачем он нужен клеткам, не до конца известно. Из-за мутации ген SMN не производит достаточного количества белка SMN, это ведёт к разрушению нейронов. Так как ген SMN очень важен, у нашей ДНК есть вторая копия этого гена — SMN2. Но, к сожалению, SMN2 производит в 10 раз меньше белка SMN.
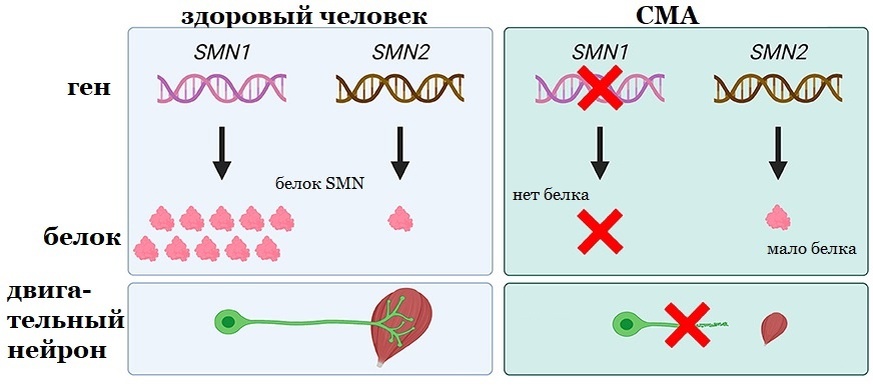
Большинство генов наследуются в количестве двух штук — один от родителя номер 1, а второй от родителя номер 2. Шучу, шучу, от мамы с папой. Так вот, у больных СМА мутации в обоих генах SMN. Если наследуется лишь один ген, только от одного родителя, то человек является носителем правильно работающего гена и мутирующего, так примерно у одного из 40 человек. Если оба родителя носители, то вероятность рождения ребёнка с СМА является 1 к 4, носителем — 2 к 4 и здоровым (не носителем) — 1 к 4.
Но иногда гены случайно дублируются, что приводит к тому, что их становится больше, чем обычных две копии. Поэтому СМА может протекать по-разному, в зависимости от того, сколько копий запасного гена SMN2 имеется. Чем больше копий, тем менее интенсивны симптомы СМА. Это также означает, что небольшое увеличение количества белка SMN может улучшить качество жизни человека со СМА.
К сожалению, СМА не лечится, но существуют методы лечения, улучшающие симптомы. Наш собственный организм затрудняет лечение СМА. Спинной и головной мозг являются самыми важными органами, поэтому они защищены особым барьером из кровеносных сосудов и определённых клеток, который не пропускает огромное количество веществ в мозг. Нусинерсен (торговое название Spinraza) был первым препаратом, одобренным для лечения СМА. Это лекарство распознает промежуточную молекулу, производимую SMN2, и повышает эффективность, с которой промежуточная молекула превращается в полноценный белок SMN. Нусинерсен необходимо вводить непосредственно в жидкость, омывающую спинной и головной мозг, чтобы преодолеть тот самый барьер. Четыре дозы препарата вводятся в первые 2 месяца лечения, а затем его вводят раз в 4 месяца.
Другой препарат — Zolgensma — является генной терапией. Здесь вирус, изменённый в лаборатории, переносит копию белка SMN1 в клетки. Вирусы имеют природную способность проникать через защитный барьер, поэтому его вводят в кровь. Оттуда он распространяется по всему организму, в том числе и попадает в нейроны. Одного укола достаточно, чтобы введённый ген был активен в течение многих лет, но мы пока не знаем точно, как долго.
Так как уколы в спинной мозг больны и проводятся только обученным персоналом в особых учреждениях, учёные разработали альтернативы. Evrysdi работает как нусинерсен, но его можно принимать ежедневно дома через рот в жидкой форме. Испытания показывают, что лучшим способом лечения СМА является начало терапии ещё до появления симптомов. Но это возможно лишь после тестирования на наличие дефектных генов SMN1 при рождении. Методы не могут полностью исцелить, но могут значительно облегчить жизнь. Стоит помнить, эти методы лечения требуют много времени, энергии и денег.
Источник:
Sleigh JN, Christie-Brown V, Ryburn L and Yáñez-Muñoz RJ (2023) Spinal Muscular Atrophy: A Rare but Treatable Disease of the Nervous System. Front. Young Minds. 11:1023423. doi: 10.3389/frym.2023.1023423
Ставя любо и подписываясь, Вы помогаете мне продвигать настоящую науку.
Предыдущая статья: Эксперимент, где всё пошло не по плану