⭐️ Синдром Хайду-Чинея (Hajdu-Cheney syndrome) – крайне редкое заболевание соединительной ткани из группы акроостеолиза. Характеризуется чрезмерной резорбцией костной ткани, что приводит к остеопорозу, развивается в сочетании с гипоплазией дуги нижней челюсти и дополнительными косточками в швах черепа.
❗️По данным литературных источников в мире было зарегистрировано около 70 случаев, с момента открытия синдрома в 1948 году.
Синдром связан с мутациями в экзоне 34, гена Notch2 (Neurogenic locus notch homolog protein 2) на хромосоме 1р12, открытым в 2011 году Симпсоном, M.A, Ирвинг. Заболевание контролируется одной парой генов, наследуется по аутосомно-доминантному типу. Однако, в большинстве описанных случаев мутация в гене выявлялась у людей без соответствующего наследственного анамнеза
В основном причина синдрома Хайду-Чинея заключена в нарушении нормального функционирования остеобластов и остаокластов.
В результате всех патогенетических изменений у людей с синдромом Хайду-Чинея развивается остеопороз и акроостеолиз. Тяжелый, плохо поддающийся лечению остеопороз приводит к компрессионным переломам (преимущественно тел позвонков), выраженному сколиозу. Больные приобретают характерный вид низкорослые, пальцы рук и ног короткие и широкие, долихоцефалическая форма черепа, широкорасставленные маленькие глаза, synophrys (широкие сросшиеся брови), низко посаженные уши, гипертрихоз.
Одним из серьезных проявлений синдрома является постепенно прогрессирующая платибазия и базиллярная импрессия, что может привести к серьезным осложнениям включая головные боли, снижение зрения, прогрессирующей гидроцефалии, нарушению дыхание, и даже внезапной смерти.
К другим проявлениям синдрома Хайду-Чинея относятся:
👍 задержка психомоторного развития,
👍 вставочные косточки черепа,
👍 недоразвитие верхней и дуги нижней челюсти, а так же лобных пазух,
👍 гипермобильность суставов;
👍 ранняя потеря зубов;
👍 снижение слуха;
👍 рецидивирующие инфекции в детском возрасте;
👍 пороки сердца;
👍 аномалии почек, такие как поликистоз.
Пациентка Р. 30 лет. в диагностический центр МРТ направлен пациент с жалобами на сильные боли в спине.
Выписка из истории болезни ФГУ «Московский НИИ Педиатрии и детской хирургии РОСЗДРАВА» детский научно-парктический центр противорадиационной защиты от 08.06.2013 г.: Хронические моторные тики. Расстройство вегетативной нервной системы по гипертоническому типу. Артериальная гипертензия. Миграция водителя ритма сердца. Дисфункция митрального клапана. Дисфункция биллиарного тракта на фоне аномалии формы желчного. Гипероксалурия. Миопия слабой степени.
Ребенок от второй беременности, протекавшей на фоне заболевания гриппом на 25 недели и миопии средней степени. Роды срочные с ранним излитием околоплодных вод. Ребенок закричал самостоятельно, оценка по шкале Апгар 9/10 баллов. Голову держит с 4 мес., сидит с 6 мес., ходит с 1 года 2 мес., говорит фразами с 3 лет. Отмечалась задержка психомоторного развития. Позднее прорезывание зубов. До года отставал в весе. Наследственные заболевания и пороки развития в родословной не наблюдаются.
Радиационный анамнез: дедушка (по линии матери) является ликвидатором аварии на "ПО Маяк" в 1958 году, получил зарегистрированную дозу облучения 383,956 мЗв. Отец ребенка проходил службу в армии на территории "ПО Маяк", доза облучения не известна. До беременности и во время мама работала на заводе рядом с "ПО Маяк".
Анамнез заболевания: отставание в физическом развитии, дисгармоничное телосложение (большая голова, короткие конечности, толстые короткие пальчики на руках и ногах), отмечался гипертрихоз на лице, задержка психо-моторного развития, позднее прорезывание зубов. В возрасте 14 лет проведена денситометрия: Остеопороз (-3). На ретгенограмме кистей рук, аномалии развития ногтевых фаланг, брадидактилия 1-5 пястных костей, все зоны роста закрыты, костный возраст соответствует 17 годам. Через год проведена повторная денситометрия: враженный остеопороз (-4,7), на фоне приема Са-Д3-никомед. Денситометрия в 20 лет: выраженный остеопороз (-7,8), на фоне лечения остеогенон и миакальцик спрей.
Объективный статус: масса тела 71 кг, рост 155 см. Окружность головы 64 см. окружность грудной клетки 89 см. Дисгармоничное телосложение (большая голова, деформация черепа грибовидного характер, череп долихоцефалической формы, в затылочной части утолщение кости). Гипоплазия нижней челюсти, неправильный рост зубов. Выраженный сколиоз грудного отдела позвоночника. Короткие конечности, брадидактилия и утолщение пальцев рук. Плоскостопие. Увеличенная подвижность в суставах. Мышечный тонус понижен. Гипертрихоз на лице и конечностях.
Цитогенетический анализ: поражены хромосомы группы С. Анализ репарационной активности ядерной ДНК: репарабельность геномной ДНК у УФ радиации снижена. Иммунологическое исследование крови: Лимфоцитоз. Активация фагоцитоза с повышенной поглотительной и оксидазной активностью нейтрофилов. Повышен уровень клеток экспрессирующих маркер апоптоза (СД95-41%). Нарушение иммунного статуса по лимфопролиферативному типу с активацией системы фагоцитов.
Пациенту проведено МРТ головного мозга, грудного и поясничного отделов позвоночника.
Выявлены следующие изменения (Рис. 1.):
A. долихоцефалическая форма черепа, с батроцефалией;
B. платибазия и базиллярная импрессия;
C. расширение швов черепа, с наличием множества мелких вставочных косточек по затылочному шву;
D. аплазия фронтальных и клиновидной пазух;
E. уплощение и преимущественно горизонтальная ориентация ската, и расширение и уплощение турецкого седла.
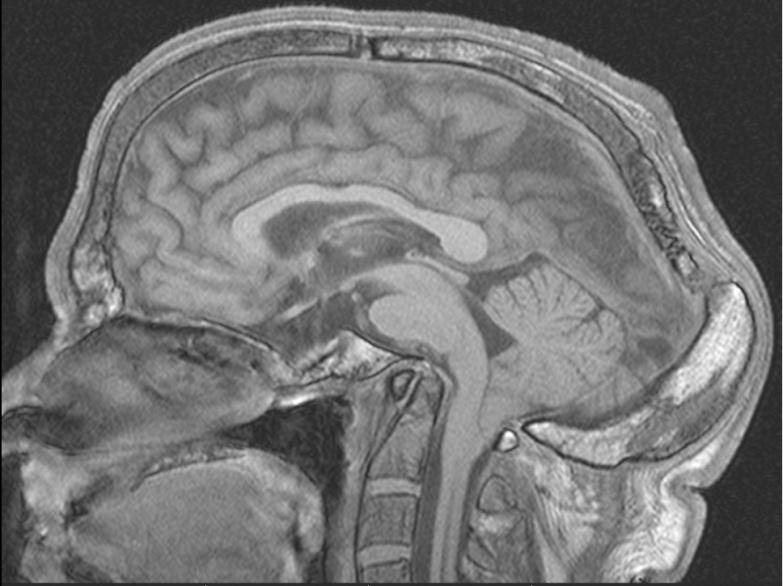
Рис.1. А, В, Е. Режим Т1_ВИ: долихоцефалическая форма черепа – увеличение сагиттального размера, и батроцефалия – утолщение затылочной кости. Уплощение и преимущественно горизонтальная ориентация ската, и расширение и уплощение турецкого седла; уменьшение краниовертебрального угла до 98гр.; увеличение угла Богарта до 166 гр. - платибазия; с. Зубовидный отросток С2 позвонка расположен выше линии Чемберлена на 2,2 см – базиллярная импрессия.
Рис.1. С. В режимах Т1_ВИ (а) и Т2 (b): а – расширение затылочного шва; b – дополнительные косточки.
Рис. 1. D. В режимах Т2 и Т1: аплазия фронтальных пазух (тонкая стрелка) и клиновидной пазухи (короткая стрелка).
В сочетании с характерными изменениями черепа и краниовертебрального перехода, у пациента выявлена аномалия Арнольда-Киари II (Рис. 2.)
Рис. 2. Аномалия Арнольда-Киари II: большое затылочное отверстие расширено; опущение миндалин мозжечка ниже плоскости большого затылочного отверстия на 1,1 см; удлинение и пролабирование ниже плоскости большого затылочного отверстия продолговатого мозга; мост сужен в сагиттальной плоскости; уменьшение в объеме предпонтинной и большой цистерны мозга; четверохолмная пластинка уплощена, вытянута в передне-заднем направлении; уменьшение понто-фораминального расстояния до 0,4 см; гидромиелия (длинная стрелка).
Гидроцефалия (внутренняя тривентрикуллярная, в сочетании с расширение субарахноидального пространства) является вторичной, вследствие выраженной дисплазии краниовертебрального перехода.
Так же были выявлены и другие сочетанные изменения: гипоплазия передних отделов левой височной доли с компенсаторным локальным расширением субарахноидального пространства /по типу арахноидальной ликворной кисты - вариант развития/; единичные очаги глиоза лобных долей /вероятнее, сосудистого генеза/.
При МР исследовании грудного и поясничного отделов позвоночника, выявлена выраженная остеопоротическая деформация тел позвонков, с наличием подострых компрессионных переломов тел Th12 и L1, L2 позвонков (Рис. 3), деформации тела Th11 позвонка (что соответствует перелому в хронической стадии). С формированием деформации осанки в виде смещения грудного кифоза вниз до уровня Th9-L2 сегментов, усиление поясничного лордоза, и S-образной сколиотической деформации исследуемых отделов позвоночника (Рис. 4). Гидромиелия на уровне С2-Th6 сегментов (Рис. 5).
Рис. 3. Подострые компрессионные переломы тел Th12 и L1, L2: МР-сигнал от тел позвонков неоднородный, с признаками умеренно выраженного отёка костного мозга (гиперинтенсивный МР-сигнал в режимах Т2и STIR, гипоинтенсивный Т1).
Рис. 4. Сколиотическая деформация грудного (а) и поясничного (b) отделов позвоночника. Смещения грудного кифоза вниз до уровня Th9-L2 сегментов.
Рис. 5. Спинной мозг неоднородной структуры за счет веретенообразной гидромиелической полости в центральных его отделах, с неравномерным истончением вещества мозга.
На основании всех совокупных данных, можно сделать вывод о наличии у пациента Синдром Хайду-Чинея.
Синдром Хайду-Чинея (Hajdu-Cheney syndrome) – крайне редкое заболевание соединительной ткани из группы акроостеолиза. Характеризуется чрезмерной резорбцией костной ткани, что приводит к остеопорозу, развивается в сочетании с гипоплазией дуги нижней челюсти и дополнительными косточками в швах черепа.
По данным литературных источников в мире было зарегистрировано около 70 случаев, с момента открытия синдрома в 1948 году.
Синдром связан с мутациями в экзоне 34, гена Notch2 (Neurogenic locus notch homolog protein 2) на хромосоме 1р12, открытым в 2011 году Симпсоном, M.A, Ирвинг. Заболевание контролируется одной парой генов, наследуется по аутосомно-доминантному типу. Однако, в большинстве описанных случаев мутация в гене выявлялась у людей без соответствующего наследственного анамнеза
В основном причина синдрома Хайду-Чинея заключена в нарушении нормального функционирования остеобластов и остаокластов.
В результате всех патогенетических изменений у людей с синдромом Хайду-Чинея развивается остеопороз и акроостеолиз. Тяжелый, плохо поддающийся лечению остеопороз приводит к компрессионным переломам (преимущественно тел позвонков), выраженному сколиозу. Больные приобретают характерный вид низкорослые, пальцы рук и ног короткие и широкие, долихоцефалическая форма черепа, широкорасставленные маленькие глаза, synophrys (широкие сросшиеся брови), низко посаженные уши, гипертрихоз.
Одним из серьезных проявлений синдрома является постепенно прогрессирующая платибазия и базиллярная импрессия, что может привести к серьезным осложнениям включая головные боли, снижение зрения, прогрессирующей гидроцефалии, нарушению дыхание, и даже внезапной смерти.
К другим проявлениям синдрома Хайду-Чинея относятся: задержка психомоторного развития, вставочные косточки черепа, недоразвитие верхней и дуги нижней челюсти, а так же лобных пазух, гипермобильность суставов; ранняя потеря зубов; снижение слуха; рецидивирующие инфекции в детском возрасте; пороки сердца; и аномалии почек, такие как поликистоз.
Несмотря на наследственный характер заболевания, в нашем случае большая вероятность экзогенного воздействия, что приводит к мутации гена, что не маловажно, для современного мира с постоянным увеличением радиационного фона. Особую важность имеет ранняя диагностика описанного синдрома для назначения поддерживающей (симптоматической) терапии, рассмотрения возможного хирургического лечения для отсрочки инвалидизации и возможной преждевременной смерти больных.
Библиографический список
- Brennan AM, Pauli RM (May 2001). "Hajdu--Cheney syndrome: evolution of phenotype and clinical problems". Am. J. Med. Genet. 100 (4): 292–310.
- A Simpson, Michael; Irving, Melita D; Asilmaz, Esra; Gray, Mary J; Dafou, Dimitra; Elmslie, Frances V; Mansour, Sahar; Holder, Sue E; Brain, Caroline E (2011). "Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss". Nature Genetics 43 (4): 303–305.
- Majewski J, Schwartzentruber JA, Caqueret A, Patry L, Marcadier J, Fryns JP, Boycott KM, Ste-Marie LG, McKiernan FE, Marik I, Van Esch H; FORGE Canada Consortium, Michaud JL, Samuels ME. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum Mutat. 2011 Oct;32(10):1114-7.
- Brown, D. M., Bradford, D. S., Gorlin, R. J., Desnick, R. J., Langer, L. O., Jr., Jowsey, J., Sauk, J. J., Jr. The acro-osteolysis syndrome: morphologic and biochemical studies. J. Pediat. 88: 573-580, 1976.
- Elias, A. N., Pinals, R. S., Anderson, H. C., Gould, L. V., Streeten, D. H. P. Hereditary osteodysplasia with acro-osteolysis (the Hajdu-Cheney syndrome). Am. J. Med. 65: 627-636, 1978.