Журнал «Менеджмент качества в медицине», март 2023
Рубрика: Обращение медизделий
Автор: Оксана Трошина

Рассматриваются ключевые моменты системы менеджмента качества, взаимосвязь инженера-конструктора с лечащим врачом при производстве индивидуальных медицинских изделий на примере жизненного цикла изделия, где основным способом производства являются аддитивные технологии.
Одно из наиболее развивающихся современных направлений медицинской промышленности связано с использованием аддитивных технологий. Применение 3D-принтеров позволяет печатать, например титановые имплантаты любой конфигурации и вспомогательный инструмент, облегчающий проведение операции.
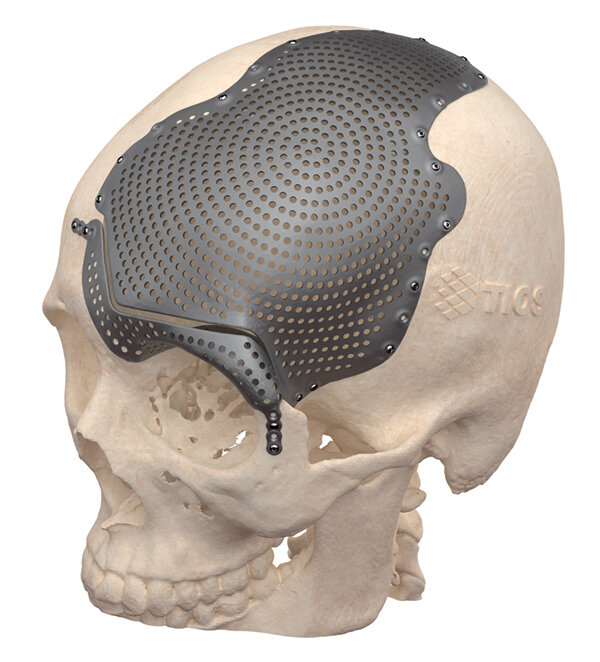
Индивидуальные титановые имплантаты изготавливаются строго в соответствии с антропометрическими данными пациента, что, несомненно, имеет ряд преимуществ. Во-первых, это позволяет обеспечивать самые разные локализации замещения: свод черепа, тазобедренные суставы со сложными резекциями, локтевые и лучезапястные суставы, мелкие кости кистей рук, пятки, кости грудной клетки. Во-вторых, такой имплантат точно заменяет резецированный участок и закрепляется непосредственно в местах, где это более всего необходимо. Помимо имплантатов, хирургам для проработки хода операции необходимы анатомические модели из пластика или других материалов, изготовленные с применением аддитивных технологий.
Индивидуальные медицинские изделия могут применяться при лечении онкологических заболеваний, например для эндопротезирования при обширных резекциях со сложной локализацией очага, в нейрохирургии, в челюстно-лицевой хирургии, ортопедии, травматологии (при первичном, а также в ревизионном эндопротезировании суставов и костей в случае развития перипротезных инфекций) (рис. 1–3).
Контроль качества производства подобных индивидуальных медицинских изделий должен быть непрерывным. Для оптимизации процесса контроля на производстве необходимо внедрить и поддерживать систему менеджмента качества (СМК), соответствующую требованиям ISO 13485 «Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования» [1]. Процессы СМК помогают осуществлять контроль каждого этапа производства медицинского изделия и таким образом выпускать продукцию, гарантированно соответствующую предъявляемым требованиям, которая не создаст риска для пациента.
В статье я хочу поделиться своим опытом внедрения СМК для контроля качества индивидуальных медицинских изделий. Опустив общие организационные моменты, все внимание переключим на специфику контроля качества процессов производства с применением аддитивных технологий.
Индивидуальный имплантат – это имплантат, который проектируется по антропометрическим данным конкретного пациента и будет подходить только ему, поэтому при производстве таких изделий важная роль отводится взаимодействию инженера-конструктора и лечащего врача. Схема взаимодействия показана на рис. 4. Лечащий врач определяет необходимость в эндопротезировании индивидуальным имплантатом и передает данные КТ в компанию-производитель.
Обработка персональных данных должна осуществляться с соблюдением принципов и правил, установленных Федеральным законом от 27.07.2006 № 152-ФЗ «О персональных данных» [2]. Из лечебной медицинской организации передаются на материальном носителе данные, полученные с применением компьютерной или магнитно-резонансной томографии (КТ или МРТ), необходимые для изготовления индивидуального изделия. К материальному носителю прилагается заявка на изготовление индивидуального изделия, подписанная лечащим врачом. Заявка должна содержать следующую информацию:
Ф.И.О. пациента;
его возраст;
рост, вес;
диагноз пациента;
данные о состоянии костей пациента;
данные о локализации замещения кости имплантатом;
тип и наименование планируемой операции;
основные желаемые характеристики медицинского изделия (при наличии).
Заявка подтверждает, что конкретный пациент действительно нуждается в эндопротезировании индивидуальным имплантатом, а его персональные данные позволят инженеру спроектировать наиболее подходящее для конкретного случая изделие. Поскольку компания-производитель работает с пациентом не напрямую, а посредством лечащего врача, она не запрашивает письменного согласия пациента на обработку его персональных данных. Ответственность за охрану персональных данных несет лечебное учреждение. Однако все персональные данные и медицинские записи в компании-производителе должны находиться в управляемых СМК условиях. Персональные данные пациента должны быть идентифицированы путем присвоения уникального номера, который сопровождает производство и обеспечивает прослеживаемость медицинского изделия на всех стадиях его жизненного цикла.
После получения данных КТ или МРТ проводится оценка качества и полноты вводной информации.
Инженер-конструктор с помощью специального лицензионного валидированного программного обеспечения (ПО) выполняет предварительную оценку возможности работы с переданными файлами, а также проверяет достаточность данных для проектирования индивидуального изделия.
Особое значение имеет срок передачи данных КТ с момента сканирования, поскольку заболевание костей у пациента может быстро прогрессировать, опухоль разрастаться, и если от сканирования методом КТ пройдет слишком много времени, выбор уровней резекции окажется некорректным. Большое значение также придается качеству снимков, их количеству и релевантности.
Далее выполняется сегментация с выделением всех элементов области интереса, составляется первичное техническое задание для проектирования изделия, определяется необходимая комплектность индивидуального имплантата, например дополнительные модели для предоперационного планирования, шаблоны для опилов и рассверливаний, примерочные имплантаты из пластика и др.
После согласования с лечащим врачом уровней резекции и необходимой комплектации эндопротеза создается 3D-модель индивидуального изделия. В соответствии с рекомендациями оперирующего хирурга инженер-конструктор с помощью соответствующего ПО в течение 2–10 рабочих дней, в зависимости от сложности модели, проектирует индивидуальное изделие для выполнения определенной хирургической задачи с учетом антропометрических данных конкретного пациента.
На этом же этапе формируется сопроводительная эксплуатационная техническая документация с описанием комплектности имплантата, основных его параметров, описанием фиксации и правил сборки. Составление сопроводительной документации должно соответствовать локальным регламентам организации.
Сопроводительная эксплуатационная документация необходима не только лечащему врачу, но и пациенту. Например, техническая документация, инструкция по применению поставляется в комплекте с эндопротезом и передается хирургу, а паспорт имплантата – пациенту.
В сопроводительную документацию необходимо включить данные проведения менеджмента риска для конкретного изделия по ГОСТ ИСО 14971–2011 «Изделия медицинские. Применение менеджмента риска к медицинским изделиям» [3]. В процессе менеджмента риска определяются опасные ситуации, которые могут произойти в ходе установки или эксплуатации изделия, определяется их тяжесть и вероятность появления, по этим данным происходит построение матрицы риска. Если риск пересекает недопустимые границы, следует запланировать и провести мероприятия по снижению его уровня, в результате чего формируется матрица с остаточным риском. Любой остаточный риск, сохраняющийся после выполнения предупреждающих мероприятий по управлению риском, нужно оценить в соответствии с установленными критериями. Если в процессе выполнения мер по управлению риском изготовитель устанавливает, что требуемое уменьшение уровня риска практически не осуществимо, он должен выполнить анализ соотношения риск – польза для остаточного риска. И если собранные доказательства свидетельствуют о том, что польза от предусмотренного применения медицинского изделия не превышает остаточный риск, то риск считается недопустимым. Однако в случае, когда польза от применения медицинского изделия превышает предполагаемый риск, можно перейти к анализу эффективности мер и описанию влияния остаточного риска в технической документации. Если остаточные риски оценены как допустимые, изготовитель должен принять решение, о каких остаточных рисках следует информировать и какую именно информацию необходимо включить в техническую документацию в целях привлечения внимания к остаточным рискам.
После того как возможный риск выявлен и сведен к минимуму, необходимо убедиться, что модель эндопротеза будет отвечать всем заявленным в техническом задании требованиям и сможет выдерживать определенную нагрузку. Валидация и верификация изделия подразумевает расчет прочностных нагрузок в специальных валидированных программах.
Достоверное прогнозирование срока службы протеза возможно только при учете его основных задач, сложной геометрической формы областей биомеханической системы и ее многокомпонентности, динамических неоднородных условий нагружения и многочисленных контактных взаимодействий. Для этого применяется конечно-элементный анализ. Требования к учету многочисленных деталей расчетной области, по сути, способствуют формированию многомиллионной системы математических уравнений, решение которой по силам только суперкомпьютерным технологиям [4]. Таким образом, после проведения расчетов прочностных нагрузок можно с уверенностью говорить об адекватном функционировании эндопротеза, отвечающего всем требованиям технического задания и технических условий.
После валидации проект направляется на окончательное согласование с лечащим врачом, который проводит проверку соответствия 3D-модели изделия плану хирургической операции.
Когда лечащий врач утверждает проект, модель запускается в производство. Сначала проводится подготовка 3D-моделей, а далее выполняется их печать посредством лазерного плавления порошка титанового сплава Ti-6Al-4V, входной контроль которого осуществляется по ГОСТ Р ИСО 5832–3-2020 «Имплантаты для хирургии. Металлические материалы. Часть 3. Деформируемый сплав титан-6 алюминия-4 ванадия», утвержден приказом Росстандарта от 04.08.2020 № 455-ст. [5]. Печать изделий происходит на 3D-принтерах в среде аргона. После печати изделие отжигается в вакуумной печи, снимается с платформы, очищается от поддержек. Далее будущий эндопротез при необходимости подвергается механической обработке, обработке на токарно-фрезерном оборудовании, пескоструйной обработке, происходит полирование с последующей технической очисткой детали и предстерилизационной очисткой. Каждый процесс осуществляется в соответствии с внутренней документацией предприятия.
С точки зрения СМК производственные помещения и производственные процессы подлежат обязательному аудиту по локальным стандартам компании. В ходе внутреннего аудита проверяется соответствие организации, обеспечения и условий технологических процессов требованиям действующих документов. Факты несоответствия установленным требованиям регистрируются в специальном журнале.
Аудиторская группа проводит анализ результатов проверок и формирует обобщенную информацию о состоянии СМК и ее элементов. Обобщенные материалы проверок рассматриваются на совещаниях с участием высшего руководства организации.
В случае выявления несоответствий руководитель аудитируемого подразделения формирует план организационно-технических мероприятий по устранению причин этих несоответствий с указанием сроков выполнения, исполнителей и ресурсов, необходимых для реализации мероприятий, при необходимости готовит и представляет руководству организации предложения по совершенствованию элементов СМК, связанных с проверяемой работой.
По результатам внутреннего аудита руководитель группы составляет отчет, который должен включать следующую информацию:
цель аудита;
объем аудита;
сведения об аудиторской группе;
дата и место проведения;
критерии аудиторской оценки;
наблюдения аудита;
заключения по результатам аудита;
рекомендации по улучшению.
Отчет должен быть датирован, подписан всеми внутренними аудиторами и согласован с руководителем. К отчету прикладывается план аудита и дополнительные материалы. Результаты аудита могут указывать на необходимость выявления причин возникновения несоответствий, а также проведения корректирующих и предупреждающих мероприятий по улучшению, которые проектирует руководитель проверяемого подразделения. Разработанные мероприятия реализуются ответственными исполнителями в установленные планом сроки. В случае невозможности проведения мероприятий в установленные сроки руководитель подразделения обязан известить об этом главного аудитора в письменной форме и согласовать их перенос на другое время.
Контроль выполнения мероприятий по устранению несоответствий, выявленных в ходе аудита, и оценку их результативности по представленным свидетельствам осуществляет руководитель аудиторской группы.
Несмотря на то что контроль изготовления индивидуального медицинского изделия происходит на каждом этапе производства, одним из основных факторов поставки качественного, полностью удовлетворяющего требованиям врача и пациента изделия служит выпускающий контроль. Именно выпускающий контроль оценивает внешний вид изделия: наличие на нем вкраплений ржавчины, заусенцев, царапин недопустимо.
Завершает этап контроля упаковка изделий. Каждая индивидуальная упаковка маркируется в соответствии с техническими условиями производителя и по требованиям национальных стандартов ГОСТ Р ИСО 14630–2017 «Имплантаты хирургические неактивные. Общие требования» [6] и ГОСТ Р ИСО 15223-1–2014 «Изделия медицинские. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации. Часть 1. Основные требования» [7]. Образец маркировки медицинского изделия разрабатывается конструктором и входит в состав конструкторской документации.
После упаковки и маркировки индивидуальные медицинские изделия проходят процесс стерилизации.
Процесс стерилизации медицинского изделия подлежит обязательной валидации, так как требуется доказать, что эта продукция действительно стерильная и упаковочное решение способно сохранить ее стерильность. Срок сохранения стерильности, указанный заявителем, должен быть подтвержден лабораторными испытаниями, например методом ускоренного старения.
Процедуры валидации обеспечивают надлежащее качество продукции и снижают риски поставки в клинику нестерильного (контаминированного) изделия.
Для контроля качества стерилизации на предприятии применяются биологические и химические индикаторы, зарегистрированные в Российской Федерации, имеющие полный комплект необходимых подтверждающих документов. Стерильность подтверждается методом посева на стерильность в аккредитованной лаборатории.
Стерилизация титановых имплантатов проводится согласно ГОСТ Р ИСО 17665-1–2016 «Стерилизация медицинской продукции. Влажное тепло. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий» [8].
Использование медицинских индивидуальных изделий должно осуществляться в соответствии с инструкцией по применению и технической документацией на конкретное индивидуальное медицинское изделие.
Использование медицинских изделий регламентируется множеством правил и ограничений, поэтому к имплантации допускается медицинский персонал, имеющий опыт работы в травматологии, ортопедии, эндопротезировании, челюстно-лицевой хирургии, детально изучивший документацию на изделие.
После установки эндопротеза жизненный цикл изделия не заканчивается, поэтому производителю важно внедрить процедуру обратной связи, которая позволяет отследить, удалось ли установить имплантат согласно документации и отвечает ли эндопротез всем заявленным требованиям. Для этого проводится сбор и анализ комментариев оперирующих хирургов, постоперационных данных КТ-исследований на соответствие выполненной операции ее плану с установленными уровнями резекции. Помимо этого, собираются и анализируются постоперационные данные более поздних периодов.
Разработка индивидуальных медицинских изделий, их проектирование, производство – сложный, многоэтапный процесс, связанный с тесным взаимодействием конструктора и врача, предусматривающий моментальную реакцию и слаженную работу всего коллектива. Внедрение СМК на производстве медицинских изделий позволит оптимизировать основные и вспомогательные процессы, поможет отслеживать медицинское изделие на каждой стадии его жизненного цикла для того, чтобы выпустить изделие, которое будетточно соответствовать всем заявленным требованиям. Процессы СМК дают возможность заблаговременно предупредить любые отклонения, которые могут привести к производству несоответствующей продукции. Политика предприятия в области качества включает принципы, задачи и обязательства компании в области качества, которые согласуются с ожиданиями врачей и запросами пациентов (рис. 5, 6).
Источники
[1] ГОСТ ISO 13485–2017 «Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования» (дата введения 01.06.2018). М.: Стандартинформ, 2018.
[2] Федеральный закон от 27.07.2006 № 152-ФЗ «О персональных данных» [Электронный ресурс] // Кодекс. URL: https://docs.cntd.ru/document/901990046.
[3] ГОСТ ИСО 14971–2011 «Изделия медицинские. Применение менеджмента риска к медицинским изделиям» (дата введения 01.01.2013). М.: Стандартинформ, 2013.
[4] ISSN 2409-6601 // Российский журнал биомеханики. 2018. Т. 22. № 3. С. 437–458.
[5] ГОСТ Р ИСО 5832-2–2020 «Имплантаты для хирургии. Металлические материалы. Часть 2. Нелегированный титан» (дата введения 01.05. 2021). М.: Стандартинформ, 2020.
[6] ГОСТ Р ИСО 14630–2017 «Имплантаты хирургические неактивные. Общие требования» [Электронный ресурс] // Кодекс. URL: https://docs.cntd.ru/document/1200144221.
[7] ГОСТ Р ИСО 15223-1–2014 «Изделия медицинские. Символы, применяемые при маркировании на медицинских изделиях, этикетках и в сопроводительной документации. Часть 1. Основные требования» [Электронный ресурс] // Кодекс. URL: https://docs.cntd.ru/document/1200110953.
[8] ГОСТ Р ИСО 17665-1–2016 «Стерилизация медицинской продукции. Влажное тепло. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий» (дата введения 01.03.2017). М.: Стандартинформ, 2016.
***
РИА «Стандарты и качество»
Тел. +7 (495) 771-66-52, пишите на e-mail: podpiska@mirQ.ru
или оставляйте заявку на нашем сайте https://ria-stk.ru/
Присоединяйтесь к сообществам издательства «Стандарты и качество»:
VK: https://vk.com/ria_stk
YouTube: https://www.youtube.com/channel/UCvW86WE6yIaFNZqK5swi70A
#СТандартыиКачество #МенеджментКачествавМедицине