После открытия функционала ДНК ученые сразу задумались об её организации. Называли библиотекой , некоторые считали , что она связана с «живой» вычислительной машиной клетки. Речь шла о Вычислительной машине Последовательного типа. Но, решить эту проблему не получалось. В 2003 году после моей первой работы по происхождению Жизни, как-то автоматически встал вопрос о следующем шаге – вплотную приблизившись к ДНК. Ясно было, что эта странная машина если уж есть, то всё держит при себе. Память и процессирующие модули. Теперь вроде уже очевидно, что комбинаторика должна была быть инструментом такого аналитического исследования. То есть, очевидно, что весь секвенс ДНК – должен быть «оцифрован». Почему? обычные вычислительные машины имеют миниум две шины - по одной идут инструкции и адреса, по другой - данные. здесь "шина - одна. И именно "тип" слов -будет "ключом" к его смыслу. Что же там за цифры ? Это могут быть только наборы из последовательностей A , T , C , G . Ясно, что эти «слова» не должны допускать двузначности. Одни - адреса, другие - инструкции, третьи - данные. То есть комбинация – набор слов. При этом, их размер должен быть связан с размером генома (ДНК).
Но параллельно уже развивалась другая идея: Эволюция двойничной (А,Т) первичной Жизни в четверичную ( C , G , A ,Т ). Потому что теория утверждала, что Жизнь зародилась двойничной и под главные кандидаты в первую пару попали А и Т(U) (аденин и тимин(Урацил)). Ведь речь идет о РНК происхождении Жизни.
И первым решением было нахождение Главного Уравнения Жизни через эту модель: распада матрицы (А,Т).
Новая пара нуклеиновых оснований ( C , G ) нашла свое место в эволюции. Она появилась позже - как модификация "А" и сначала просто была "вписана" в "инструменты" для разделения информации (А и Т), это потом появится полноценный синтез этой пары (С и G) и появится механическая возможность (достаточность материала) для полноправного участия в шифровании ДНК. Ясно, что мы находимся далеко от этой удивительной эпохи – БИНАРНОЙ ЖИЗНИ . С другой стороны, мы можем наблюдать сложные эволюционные процессы:
1. Распад `AТ` платформ.
2. Замещение `AТ ` платформ - `CG` платформами.
3. Вырождение `AТ` - платформ.
4. Вырождение `CG` - платформ.
Распад матриц мы можем определить из следующих соображений:
Примем: Скорость распада мультиплета размера (N) - зависит от его связанности с геномом:
(1) dN / dt = F(N)
Где: N – размер мультиплета.
F(N) - функция зависящая от `N`- размера мультиплета и его `связанности` с геномом.
t - `нормализованное` время.
Скорость распада мультиплетов также (n) пропорциональна их количеству:
(2) dn(N) / dt = Kn * n(N)
Где: n(N) – количество мультиплетов с `N` – размерами
Kn – коэффициент.
Используя уравнения (5) и (6) получаем:
(3) dN = F(N) * dn/(( Kn )* n(N) )
Функция F(N) – зависит от степени участия мультиплетов в жизненно-важных кластерах: как `ключей`, `инициирующих последовательностей` и других знаков в `грамматике` генома. Можно ожидать три простейших случая:
(4) а) F(N) = KN * N - когда мультиплеты размера -`N` не связаны своим размером с функциями генома;
Где: KN – Коэффициент характеризующий связь мультиплетов `N` размера с геномом (связь слабая);
б) F(N) = KN – когда мультиплеты `N` размера связаны своими кодирующими свойствами с геномом;
в) F(N) = KN /N - когда мультиплеты размера `N` сильно связаны своим размером с функциями генома и их оптимальная для генома мутация пропорциональна их размеру;
Соответственно, получаем основные зависимости `N` от` n(N)`:
(5) для 4 а Ln ( Ni/Nk ) = K1 * Ln ( n(Ni) / n(Nk) )
(6) для 4 б Ni -Nk = K1 * Ln (n(Ni) / n(Nk) )
(7) для 4 в Ni^2 -Nk^2 = K1 * Ln (n(Ni) / n(Nk) )
Где: К1 = KN / Kn
Можно ожидать, что разные размеры и виды мультиплетов будут находиться в различной зависимости от генома и, соответственно, описываться различными уравнениями. Эти зависимости позволяют измерять `дискретное` значение скорости мутаций в отдельных участках кластеров геномов . Ясно, что эти значения носят вероятностный характер и требуют дополнительных подтверждений.
Из секвенсов ДНК разных организмов зависимость (6) является глобальной. Исследовались сотни геномов из разных Царств Живых организмов, все они показали приверженность к этой модели. Только сложность и размер генома усложняли эти зависимости на высоких значениях размеров «слов».
Так граф 1 - Ehctericia Coli O 157 H 7, В – Encephalitozoon cunculi (хромосома 4), С – Giardia lamblia (хром. 1). Везде ось У - Ln(n) , а ось Х - Ni.
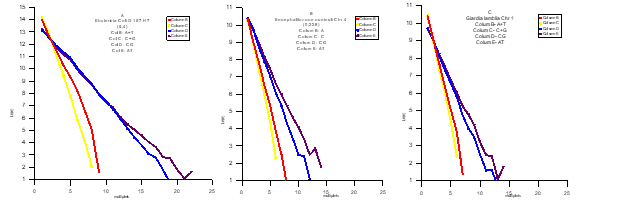
Более того, оказалось , что хромосомы являются тождественными отображениями генома. Это было поразительно и ,конечно, послужили главным и прецизионным доказательством истинности модели (пример граф 2 – Drosophila melanogaster (геном – 2 R ,2 L ,3 R ,3 L , X ,4)
Граф 2 Drosophila melanogaster ( геном – 2R,2L,3R,3L,X,4)
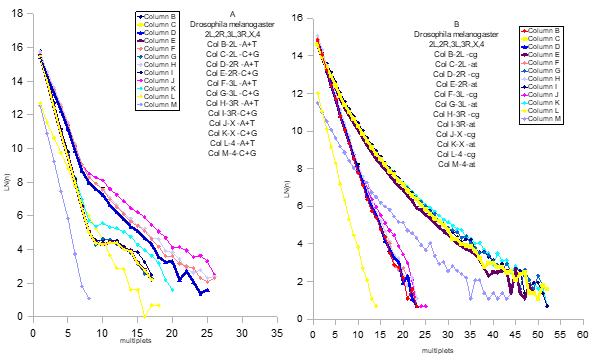
Интересно, что мы впервые видим в генетике прямые линии, а не облака из точек. То есть, процессинг действует очень жестко, с математической точностью.
Второй способ решения этой задачи через комбинаторику дал тот же результат:
Рассмотрим следующую проблему в развитии генома (родственную проблеме в информационных технологиях) – которую назовем ‘оптимизацией кода’. Чтобы обеспечить 'гладкий' процесс развития через мутации (которые неизбежно последуют мультиплицирование в геноме), геном, при наращивании своей длины, должен был оптимизировать длину ‘адресного слова’. При слишком длинном адресном векторе, процесс их 'подгонки' 'присвоение нового адреса' резко замедляется, так как новый ген мешает работе старого, а старый новому. Обратно, при слишком короткой длине, слишком легко, в разных местах генома, в процессе мутации, появляются 'нелегальные' адреса: что резко усложняет работу генома, его адаптацию и его эволюцию. По этой причине, геном вынужден 'решать' проблему оптимизации кодирования.
Можем записать следующее уравнение для всей информации в геноме:
1. M = B * A^B (A^B – информационный размер генома)
Где: M – физический размер генома; ^ - обозначение верхнего регистра- степени
A - код (мерность системы - скажем 4 - для A,T,C,G);
B - максимальный физический размер адресного слова (AV) .
Рис 3 - "Оцифровка всей длины ДНК на разные мультиплеты длинной "В""
Оптимум этого общего уравнения будем искать при условии: B*A = Const (которое соответствует информационному размеру AV). Смысл этого условия - ЭТО ИНФОРМАЦИОННАЯ ЕДИНИЦА. Тогда:
2. B * A = Const
Решая уравнение 1, при условии 2 получим:
3. A= e^(1-1/B)
То есть, при увеличении размера генома, ‘А’ – стремится к ‘е’.
Тогда:
4. M = B * e^(B-1)
Или
5. Ln ( M / B ) = ( B -1) где « n » = М/В
Тогда имеем :
6. Ln(n) = (B-1)
Ещё одним поразительным подтверждением этой модели была её проверка на организации триплетов в синтезе пептидов.
Синтез пептидов
Если рассмотреть начальный период развития жизни, то проблема 'оптимизации' встретилась и на этапе появления белкового синтеза. Повторяющиеся 'коды' белковых последовательностей заставили RNA-DNA систему разработать 'оптимизированный код' для оптимизации и эффективной работы механизма синтеза белков. И, видимо, все еще существует 'альтернативный синтез белка ' (RNA Driving Peptides 'RNA-DP') исполняемый только RNA (видимо подобными ncRNA или rRNA + ионами металлов (видимо: Ca, Mg) и АТФ -АДФ- АМФ. Такой синтез, наверное, работал для основных 'древних' аминокислот, предположительно кодируемых только A и U(T) основаниями:
AAA - Lisine , AAU-Asparagine, AUA-Isoleucine, AUU- Start, UUU-Phenylalanine, UUA- Leucine, UAU-Tyrosine, UAA-Stop, ( предположительно и UCU(A)-Serine, CCU(A)-Proline, CAU-Histidine, CAA-Glutamine, ACU(A)- Threonine.
Для случая промежуточного 'кодирования' пептидов, уравнение (4) опять будет справедливо, только код будет состоять из трех нуклеотидов четверичной системы (A,U,C,G).
6. Np = e^Bc
где: Np – число кодируемых функций.
Bc = 3
Получаем: Np ~ 20,1 , по таблице кодонов имеем: 20 аминокислот + 6 дополнительных функций, что намного ближе к истине, чем простое: 4^3 = 64.
Что следует из всего этого? Теория – вещь сложная, но она позволяет видеть истоки и законы управляющие явлениями. Из этих представлений оказалось возможным вычислять местные коэффициенты мутации, что практически невозможно экспериментальными методами.



















