Синдром Олбрайта — генетическое заболевание невыясненной этиологии, поражающее весь организм человека. Патология возникает сравнительно редко и проявляется асимметричной гиперпигментацией кожи, гормональным дисбалансом, полиостотической фиброзной дисплазией костей. Этот недуг относится к редким формам гонадотропнозависимого раннего полового созревания.
Синдром Олбрайта развивается в основном у представительниц прекрасной половины человечества. Он обусловлен спонтанной мутацией гена в процессе эмбриогенеза и не передается по наследству. Диагноз ставят детям возрасте 5-10 лет на основании признаков, характерных для данного заболевания
Впервые клиника синдрома была описана в 1907 году ученым Олбрайтом, в честь которого он и получил свое название. Ученый наблюдал за низкорослыми и коренастыми девочками с круглым лицом и короткой шеей. У них были обнаружены мышечные спазмы, скелетные деформации, частичное отсутствие зубной эмали, отставание в умственном развитии, признаки ожирения. Эндокринопатия проявлялась ранним половым созреванием: появлением менструаций, активным ростом грудных желез, появлением волос на лобке. Происхождение патологии Олбрайт объяснял резистентностью почечного эпителия к паратгормону, приводящей к ускоренному обратному всасыванию фосфора и развитию гиперфосфатемии. При этом у больных паращитовидные железы имели нормальное строение, а гуморальные изменения отсутствовали.
В официальной медицине синдром получил название псевдогипопаратиреоз. Нарушения фосфорно-кальциевого обмена приводят к патологии костной системы. Подобные расстройства легко объяснить устойчивостью тканей к эндогенному паратгормону. У больных отмечается отставание в психофизическом развитии. При этом отсутствует реакция на введение экзогенного паратгормона.
Больным с синдромом Олбрайта требуется консультация специалистов в области гинекологии, эндокринологии, лучевой диагностики, офтальмологии, травматологии и ортопедии.
Этиопатогенетические факторы
Этиология и патогенез патологии в настоящее время до конца не изучены. Предполагают, что синдром Олбрайта — результат спонтанных мутаций, возникающих беспричинно. У больных обнаруживают единичные дефекты генов в некоторых клетках организма. Выраженность клинических признаков патологии зависит от количества клеток, имеющих мутировавший ген. Подобные мутации случаются на постзиготной стадии эмбрионального развития, и не передаются по наследству.
Факторы, способствующие развитию синдрома:
- врожденные аномалии гипоталамуса,
- психогенные факторы — стрессы, конфликты, неврозы,
- эндокринопатии,
- гиперостоз основания черепа,
- преждевременная секреция гонадотропинов.
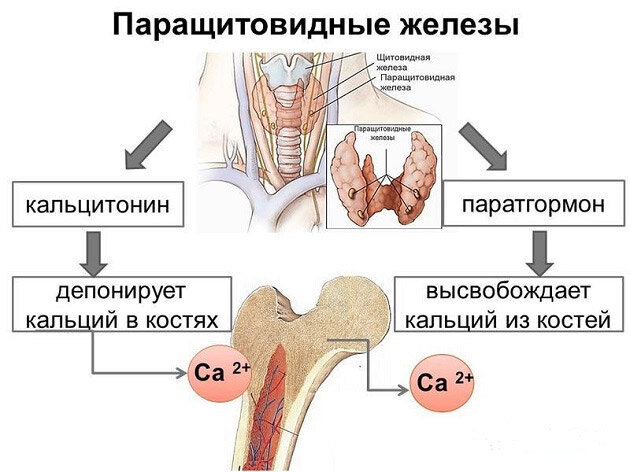
Синдром Олбрайта — результат нарушенной работы паращитовидных желез. Их основной функцией является регуляция фосфорно-кальциевого обмена. При данной патологии железы плохо реагируют на колебания этих элементов или совсем не воспринимают их.
Генетически детерминированная устойчивость структурных элементов почек и скелета к паратгормону обусловлена поражением циторецепторов или дисфункцией аденилатциклазы. Мутация специфических генов приводит к подавлению продукции цАМФ, участвующей в метаболизме, происходящем под воздействием паратгормона.
Причины дефицита цАМФ различны:
- повреждение нуклеотидсвязывающих белков клеточных мембран,
- дефект циторецепторов эпителия,
- нарушение работы аденилатциклазы.
Эти патологические изменения делают периферические ткани интактными к гормону паращитовидных желез. Органы-мишени теряют чувствительность к паратгормону, что приводит к повышению в крови уровня фосфора и снижению концентрации кальция.
Пористость костей и кистообразование — характерные изменения, обнаруживаемые при данном синдроме. Кальций вымывается из костей и откладывается в виде кальцинатов в подкожно-жировой клетчатке, мышцах, внутренних органах, эндотелии артериальных сосудов. Со временем костная ткань замещается фиброзной. Скелетные аномалии нарушают мобильность больных и становятся причиной сильной боли. Снижение витамина D в крови приводит к нарушению процесса всасывания кальция в кишечнике. Гипокальциемия стимулирует еще большее выделение в кровь паратгормона и усиленное вымывание кальция из костей, что приводит к развитию выраженной остеомаляции. Костные нарушения при этом заболевании считаются самыми тяжелыми.
На коже больных появляются пигментные пятна коричневого цвета с неровными краями. Эндокринные нарушения при данном недуге представлены ранним половым созреванием и появлением вторичных половых признаков.
Симптоматика
Синдром Олбрайта проявляется признаками поражения костной ткани, кожи и симптомами эндокринных расстройств.
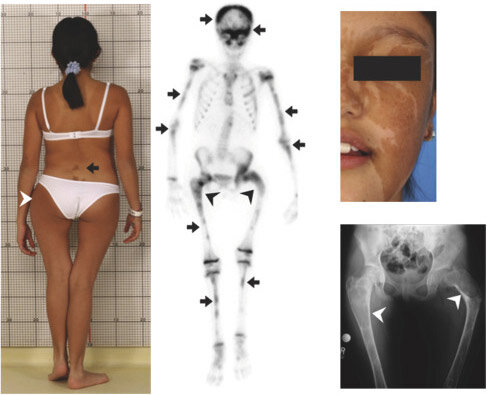
- У больных развивается фиброзная остеодисплазия, проявляющаяся различными деформациями и скелетной кривизной. Чаще всего поражаются трубчатые кости на одной конечности, что приводит к хромоте и деформации позвоночника. Уже в раннем возрасте нарушается формирование скелета. У пациентов выявляется деформация челюсти и черепа: лицевые кости утолщаются, становятся широкими, грубыми и крупными. Так формируются внешние уродства: асимметричное лицо и односторонний экзофтальм. Затем появляются симптомы артрита. Фиброзно-кистозная дисплазия представляет собой фокальное замещение кортикального слоя кости пролиферирующими фибробластами. Образование кист приводит к деформации кости и возникновению переломов. Чаще всего встречается деформация бедра по типу «пастушьего посоха».
- На коже больных появляются участки гиперпигментации, представляющие собой скопления пятен неправильной формы с неравными краями. Окраска пятен часто варьируется от желтоватой до темно-коричневой. Очаги поражения локализуются на ногах, бедрах, туловище. Пятна имеют достаточно большие размеры и гладкую поверхность. Они присутствуют на коже с рождения или распространяются по мере роста ребенка. Эпидермис при этом не изменяется. Их интенсивный рост требует гистологического исследования и оперативного вмешательства.
- Эндокринные нарушения обусловлены дисфункцией структур гипоталамо-гипофизарной области и желез внутренней секреции, расположенных на периферии. Половое созревание начинается у полугодовалых детей и заканчивается уже в семь лет. У больных начинаются месячные, развиваются молочные железы, появляются вторичные половые признаки – рост груди и появление волос на лобке. Во время ультразвукового исследования яичников в них обнаруживают крупные фолликулярные кисты. При этом сами яичники остаются нормальных размеров.
- У больных наблюдаются тонические судороги, возникающие сами по себе или под воздействием эндогенных и экзогенных раздражителей — стресса, гипо- или гипертермии, чрезмерного перенапряжения.
- Кальцинаты в коже и мягких тканях часто воспаляются и изъязвляются, доставляя больным дискомфорт.
- Возникают остеохондромы, хондроматоз, субпериостальная резорбция мелких костей пальцев рук.
- Типичные внешние признаки – низкий рост, лунообразное лицо, избыточная масса тела, короткие пальцы.
- Избыток фосфора нарушает усвояемость магния, вызывая мигрень, аритмии, боли в спине.
- Нарушается конфигурация и подвижность суставов, формируются ложные суставы.
- К прочим признакам синдрома относятся: рвота, гематурия, помутнение хрусталика, частичное отсутствие зубной эмали.
- Неврологические нарушения обусловлены атрофией зрительного нерва, потерей слуха и зрения.
- Задержка психомоторного развития.
Выделяют атипичную или бессимптомную форму псевдогипопаратиреоза, при которой количество кальция и фосфора в крови остается нормальным, отсутствуют судороги и остеомаляция.
Сопутствующие заболевания и осложнения
Синдром Олбрайта отличается не только резистентностью почек и скелета к паратгормону, но и других органов к соответствующим гормонам — яичников к гонадотропному, щитовидной железы к тиреотропному, надпочечников к антидиуретическому. Синдрому Олбрайта сопутствуют следующие эндокринопатии: гипертиреоз, акромегалия, синдром Иценко-Кушинга.
- Дисфункция щитовидной железы проявляется так называемым зобом, узелками и кистами органа.
- Избыточная продукция гипофизом гормона роста приводит к развитию акромегалии. Черты лица становятся грубыми, руки и ноги длинными, возникает артрит.
- Чрезмерная секреция гормонов надпочечников становится причиной ожирения, прекращения роста и кожной хрупкости — типичных признаков синдрома Кушинга.
Также отмечается повышенная частота аутоиммунных заболеваний, сахарного диабета, сосудистых пороков, хрящевой остеодисплазии.
К осложнениям синдрома Олбрайта относятся:
атрофия зрительного нерва, экзофтальм, патологические переломы, деформация костей, озлокачествления фиброзных фокусов, кровоизлияния в различных частях организма, кровотечения, нарушение работы кишечника, печени, почек, пищеварительной системы.
Диагностика
Диагностика синдрома начинается с выслушивания жалоб и тщательного осмотра больного. Клинические признаки настолько специфичны, что поставить правильный диагноз специалистам не составляет особого труда. Если у младенцев выявить все признаки просто невозможно, то у детей 5-10 лет легко обнаруживается характерная симптоматика.
- После изучения клинических проявлений синдрома больных направляют на генетическое исследование.
- Рентгенодиагностика позволяет обнаружить множественные пороки развития костной системы и деформации суставов.
- Лабораторные методы исследование включают анализы на различные гормоны.
- В крови обнаруживают снижение уровня кальция и повышение уровня фосфора, щелочной фосфатазы.
- Гинекологическое исследование и УЗИ органов малого таза необходимы для подтверждения изменений в яичниках.
- Специфическим диагностическим методом является тест на фосфаты и цАМФ в моче. Его проводят всем лицам с подозрением на синдром. Больным внутривенно вводят паратгормон, а спустя 4 часа собирают мочу на анализ. Если уровень гормона остается прежним, значит, имеется устойчивость ткани почек к паратгормону, и диагноз синдрома подтверждается.
Всем больным с синдромом Олбрайта требуется консультация эндокринолога, гинеколога, ортопеда — травматолога.
Лечение
Синдром Олбрайта — неизлечимое заболевание. Для поддержания жизни больных на оптимальном уровне и устранения основных клинических признаков болезни проводят симптоматическую терапию. Если общетерапевтические мероприятия не будут проведены вовремя, задержка умственного развития только усугубится.
Индивидуально разработанная диетотерапия со сниженным содержанием фосфора в рационе способствует формированию нормального уровня кальция в крови.
Комплексное лечение проявлений синдрома включает:
- препараты кальция – «Кальций Д3 Никомед», «Кальцемин», «Кальцемин Адванс»;
- препараты, связывающие фосфор и замедляющие его всасывание кишечником – «Ацетат кальция», «Нефросорб», «Ренацет»;
- средства, содержащие витамин Д – «Кальцитонин», «Миакальцик», «Сибакальцин»;
- антиандрогены – «Диане-35», «Жанин», «Ярина»;
- препараты прогестерона – «Утрожестан», «Дюфастон»;
- лекарства, подавляющие синтез гормонов щитовидной железы – «Тиамазол», «Мерказолил»;
- средства, уменьшающие синтез кортизола – «Бромкриптин», «Трилостан», «Митотан»;
- препараты, подавляющие выработку гормона роста – «Соматулин», «Октреотид», «Сандостатин»;
- ингибиторы ароматазы – «Аромазин», «Эгистразол», «Эстролет»;
- противосудорожные препараты – «Глюконат кальция», «Хлористый кальций».
Для успешного выведения избытка фосфора из организма хорошо зарекомендовал себя ежедневный гемодиализ.
Хирургическое лечение проводится при акромегалии. Также оно показано больным с прогрессирующим ухудшением зрения, выраженным болевым синдромом и серьезными увечьями. Для исправления врожденных или приобретенных дефектов костей проводят костную пластику. Опухоль или кисту из кости выскабливают, не затрагивания здоровые ткани.
Если рациональное лечение начато вовремя, прогноз для жизни благоприятный.
Комплексная и правильная терапия дает вполне благоприятный результат. Поскольку синдром Олбрайта является генетически обусловленным заболеванием, всем лицам, планирующим беременность, показано медико-генетическое консультирование. Это единственный способ профилактики данной болезни.