Болезнь Вильсона—Коновалова (гепатоцеребральная дистрофия) — редкое аутосомно- рецессивное наследственное заболевание, в основе которого лежит генетически обусловленное нарушение обмена меди с избыточным (токсичным) ее накоплением преимущественно в печени и нервной системе. Больные дети рождаются у пары родителей, оба из которых клинически здоровы, но являются носителями патологического гена. Как правило, родители не имеют случаев аналогичного заболевания в родословной. Заболевают только те индивидуумы, которые унаследовали два мутантных гена, т.е. по одному от матери и от отца — гомозиготные носители мутации. Лица, которые от одного из родителей получили мутантный ген, а от другого — нормальный ген, являются гетерозиготными носителями мутации. Заболевание у них не развивается, хотя при биохимическом исследовании могут быть обнаружены субклинические изменения в метаболизме меди.
Первое описание клинической картины болезни было сделано в 1912 г. проживающим в Англии американским неврологом Вильсоном (S. Wilson), который охарактеризовал ее как «прогрессирующая лентикулярная (лат. lenticularis — чечевицеобразный) дегенерация: семейное заболевание нервной системы, сочетающееся с циррозом печени». Вильсон впервые описал характерные изменения в головном мозге, установил постоянное наличие у больных цирроза печени. В качестве основных симптомов заболевания были отмечены разнообразные непроизвольные движения в конечностях и туловище, мышечная ригидность, приводящая к скованности, дисфагия и дизартрия, аффектные вспышки, иногда психические расстройства при отсутствии признаков поражения пирамидных путей. За 30 лет до Вильсона К. Вестфалем (Westphal C., 1883) и А. Штрюмпеллем (Strumpell A., 1898) было описано заболевание, которое по клиническому сходству с рассеянным склерозом получило название «псевдо- склероз». Заболевание характеризовалось распространенными, размашистыми, ритмичными непроизвольными движениями, повышением мышечного тонуса, амимией, дизартрией и выраженными психическими нарушениями вплоть до слабоумия. В дальнейшем оказалось, что прогрессивная лентикулярная дегенерация и псевдосклероз являются разными формами одного и того же заболевания, которое Галль (1921) назвал гепатолентикулярной дегенерацией. Тогда же выяснилось, что изменения в мозге не ограничиваются лентикулярными ядрами и нередко бывают даже силь- нее выражены в других отделах мозга. В 1953 г. Bearn, проведя анализ 30 семей, где были пациенты с болезнью Вильсона, установил аутосомнорецессивный тип ее наследования.
Представления о патофизиологии, патогенезе и клинике этой болезни были значительно расширены отечественным неврологом академиком АМН СССР Н.В. Коноваловым, который предложил название «гепатоцеребральная дистрофия», выделил 4 формы поражения нервной системы и одну абдоминальную. В 1956 г. Walshe продемонстрировал хелирующий эффект препарата D-пеницилламина. В 1974 г. Frommer привел доказательства нарушения процесса билиарной экскреции меди при болезни Вильсона. В 1985 г. Frydman и соавт. был открыт мутантный ген, детерминирующий развитие этого заболевания, а также установлено, что ген ATP7B, мутации которого вызывают заболевание, расположен в длинной части 13-й хромосомы (участок 13q14q21). В настоящее время идентифицировано более 200 мутаций гена ATP7B, которые приводят к нарушениям билиарной экскреции меди и к накоплению этого микроэлемента сначала в печени, а затем и в других органах и тканях. В результате возникает токсическое поражение органов и нарушение их функций. Именно множе- ством генных мутаций объясняются различия степени нарушений транспорта меди, клинической картины и биохимических данных в семьях больных болезнью Вильсона—Коновалова (БВК).

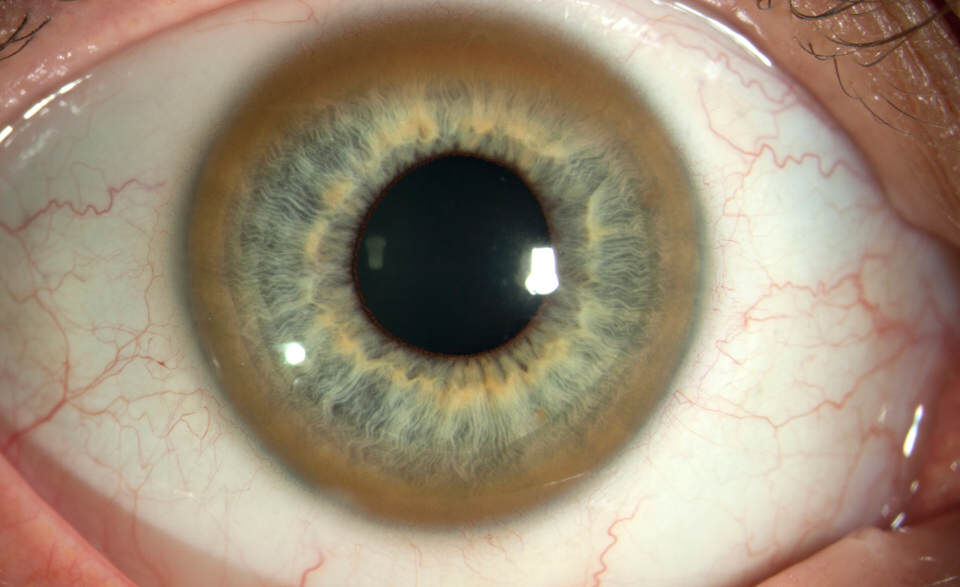
Заподозрить раннюю стадию болезни можно на основании следующих признаков: перенесенной желтухи; повторных кровотечений из носа, кровоточивости десен либо множественных кровоподтеков; сосудистых “звездочек” на коже груди и спины; своеобразных “полосок” (белых, меняющих периодически окраску на красновато-синюшную) на бедрах и в подмышечных областях; гормональных нарушений в виде аменореи или дисменореи у девушек, гинекомастии (нагрубание грудных сосков) у юношей, а также акромегалии(увеличение носа, подбородка, утолщение губ); снижения интеллекта и изменений психики в виде чередования дурашливости и пониженного настроения, трудностей усвоения нового материала, проблем с успеваемостью в школе, кольцо Кайзера-Флейшера - желтовато-зелёная или зеленовато-коричневая пигментация по периферии роговицы на десцеметовой мембране (патогномоничный симптом болезни Вильсона-Коновалова).
Гепатолентикулярная дегенерация может начать проявляться в детском, подростковом, юношеском, зрелом возрасте и очень редко – в 50–60 лет. Чем раньше начинается заболевание, тем тяжелее оно протекает (при отсутствии лечения). Однако болезнь Вильсона–Коновалова – редкий пример наследственного нарушения, для которого разработаны высокоэффективные методы лечения: даже при появлении тяжелой неврологической симптоматики систематическое лечение обычно дает “драматический” эффект, вплоть до исчезновения всех симптомов или резкого их уменьшения. Пациенты вновь могут полностью обслуживать себя, вести домашнюю работу, учиться, работать по профессии, создать семью и родить здорового ребенка. Пациентам с гепатолентикулярной дегенерацией необходимо регулярно наблюдаться у постоянного лечащего врача.
В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин.
Эти препараты назначаются по специальной схеме с постепенным увеличением дозы. К сожалению, в силу необходимости проведения пожизненного лечения и особых требований к химической чистоте препаратов отечественный аналог пеницилламина не может быть рекомендован при гепатолентикулярной дегенерации из-за высокой токсичности.
При длительном многолетнем приеме D-пеницилламина у некоторых больных гепатолентикулярной дегенерацией возникают побочные явления в виде дерматитов, анемии и иных осложнений. Поэтому был предложен альтернативный метод лечения солями цинка (оксид, сульфат и др.). У больных в пресимптоматической стадии достаточно лечения только препаратами цинка.
В настоящее время за рубежом в тяжелых случаях болезни, не поддающихся консервативному лечению, широко применяется пересадка печени. При удачном исходе операции больной полностью выздоравливает и не нуждается в дальнейшем приеме препаратов. В России делаются первые шаги в этом направлении, и одним из таких шагов является разработанный Институтом трансплантологии и искусственных органов метод биогемоперфузии с изолированными живыми клетками печени и селезенки – так называемый аппарат “вспомогательная печень”.
Помимо этих методов, большое значение имеет гепатопротекторная терапия, направленная на максимальное улучшение функций печени.
Таким образом, при правильной терапии гепатолентикулярной дегенерации – тяжелейшего наследственного заболевания мозга и внутренних органов – в 80% случаев возможно клиническое выздоровление либо выраженное улучшение состояния больных при условии своевременной максимально ранней диагностики.