Атомы – это маленькие квантовые миры, где положительно заряженные ядра окружены негативно заряженными электронами. Когда несколько таких атомов объединяются в молекулу, электроны начинают вести себя еще более запутанно. Именно поэтому компьютерное моделирование молекул считается одной из самых сложных задач в современной науке. Но, как говорится, нет ничего невозможного!
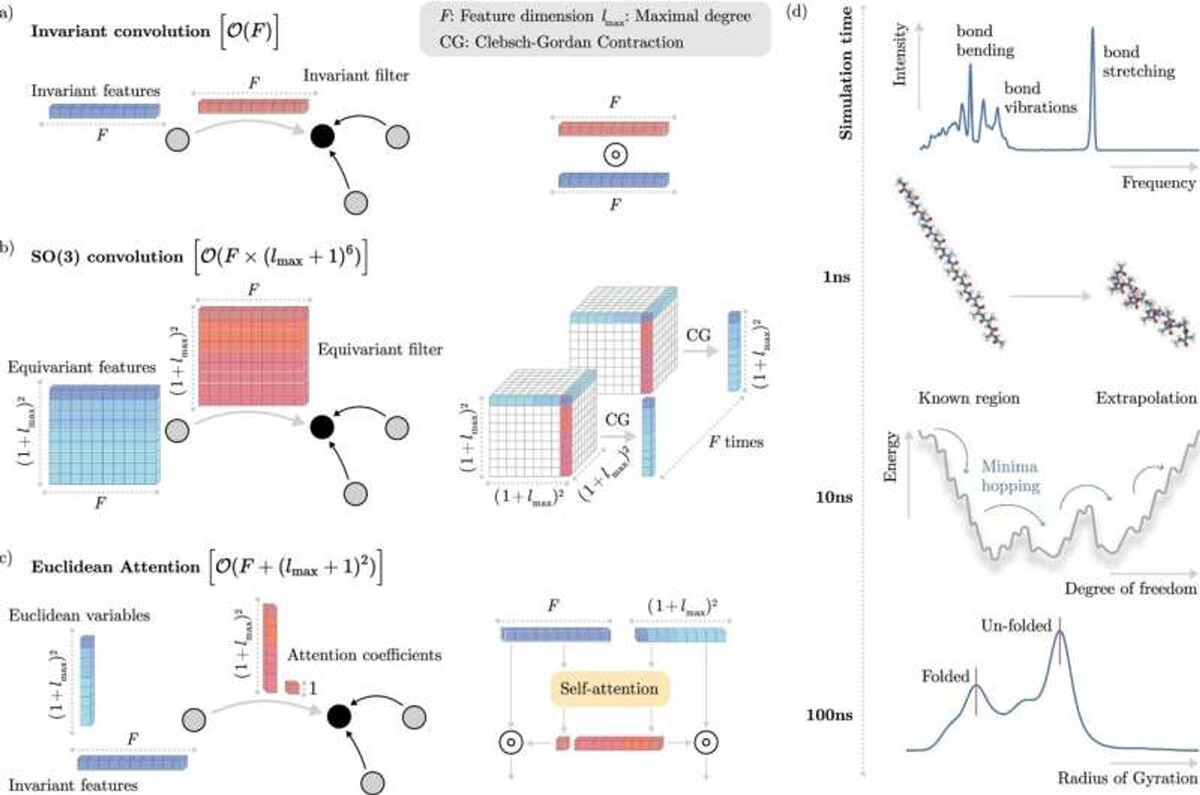
Учёные из Берлинского института основ обучения и данных (BIFOLD) при Техническом университете Берлина и Google DeepMind разработали новейший алгоритм машинного обучения, способный с высокой точностью моделировать динамику одной или нескольких молекул на длительных временных промежутках. Их работа недавно была опубликована в журнале Nature Communications.
Эти так называемые молекулярные динамические симуляции помогают понять свойства молекул и материалов, что особенно ценно для разработки лекарств и создания новых материалов, например, для солнечных батарей и аккумуляторов. Традиционные методы для вычисления взаимодействий электронов основываются на решении уравнения Шрёдингера – математического монстра, описывающего энергетические уровни квантовой системы, такой как атомы или молекулы.
Решение уравнения Шрёдингера занимает невероятное количество времени и ресурсов, особенно когда нужно моделировать взаимодействия в молекуле с десятками атомов. Представьте себе: для длительных молекулярных динамических симуляций уравнение нужно решать тысячи или миллионы раз! Даже самые мощные компьютеры начинают "пыхтеть" от таких расчетов.
"Моделирование таких взаимодействий и предсказание сложных процессов, как сворачивание белков или связывание молекул, – долгожданная мечта многих химиков и материаловедов. Это спасло бы от множества дорогостоящих и трудоемких экспериментов," – поясняет исследователь BIFOLD Торбен Франк.
Однако, благодаря методам машинного обучения эта мечта становится явью. Вместо того чтобы решать уравнение Шрёдингера в лоб, алгоритмы машинного обучения могут "научиться" предсказывать итоговые результаты электронных взаимодействий на атомарном уровне, значительно сократив вычислительную нагрузку.
Весь фокус теперь заключается в разработке эффективных алгоритмов, которые "учат" систему машинного обучения, как взаимодействуют электроны, не моделируя их явно. Многие алгоритмы используют то, что физические системы следуют определенным инвариантностям – то есть некоторые свойства молекул остаются неизменными при перемещении молекул в пространстве. Но включение этих инвариантностей в модели сложное дело и, в конечном счете, ограничивает скорость выполнения симуляций.
Исследователи из BIFOLD решили эту проблему, разработав новый алгоритм, который на начальном этапе отделяет инвариантности от другой информации о химической системе. Это позволяет модели сосредоточиться на самых сложных операциях, связанных с физической информацией, что существенно снижает общую вычислительную нагрузку.
"Симуляции, которые требовали месяцев или даже лет вычислений на мощных компьютерных кластерах, теперь могут выполняться за несколько дней на одном компьютерном узле," – говорит исследователь BIFOLD, доктор Стефан Чмела.
Как отметил профессор доктор Клаус-Роберт Мюллер, сопредседатель BIFOLD и ведущий научный сотрудник Google DeepMind, "Эта работа демонстрирует потенциал сочетания передовых методов машинного обучения с физическими принципами для преодоления многолетних проблем в вычислительной химии."
Источник:
DOI: 10.1038/s41467-024-50620-6
-------------------------------------
Поддержите наш проект: подпишитесь на канал, поставьте лайк или напишите комментарий, а также подписывайтесь на наши страницы на других площадках, в том числе на сервисе поддержки авторов Бусти. Ссылки найдёте в описании канала. Заранее спасибо!



















