Именно таким образом можно назвать то, что происходит с людьми которым поставлен диагноз - прогерия. Это редкое заболевание, которое может наследоваться по аутосомно-рецессивному признаку от родителей или возникать вследствие спонтанной мутации. Сейчас в мире зарегистрировано больше 350 случаев данного заболевания. Прогерия делится на 2 основным подтипа:
- синдром Вернера - заболевание, которое диагностируется у уже взрослых людей;
- синдром Гетчинсона-Гилфорда,врожденный тип заболевания;
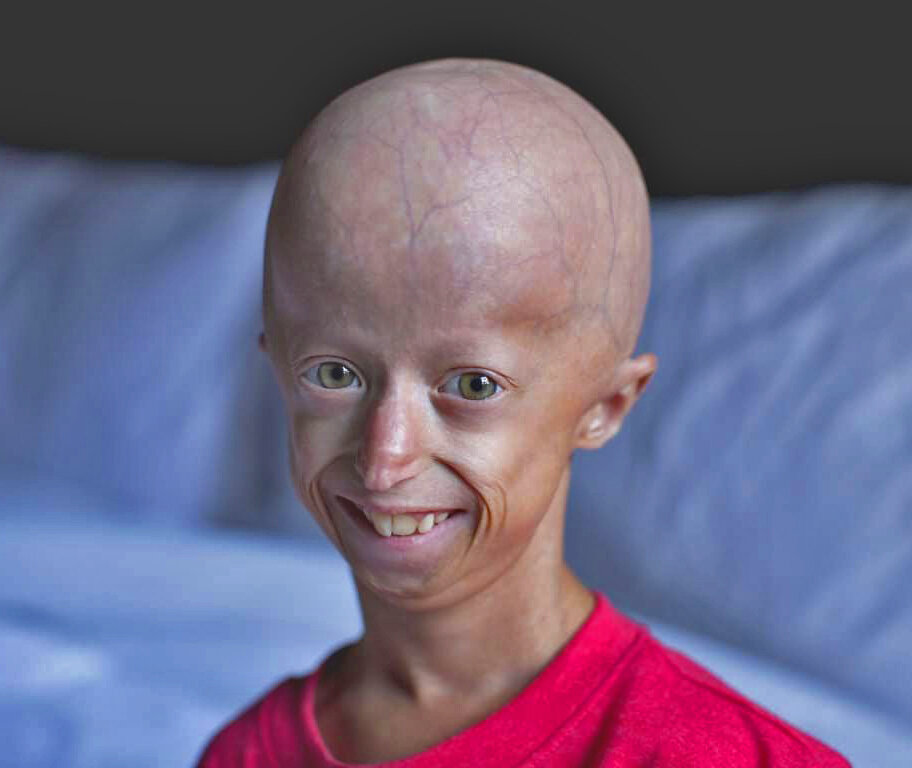
Прогерия у детей
Как правило, врожденный тип данного заболевания встречается гораздо чаще. Средний возраст до которого доживают люди с данным заболеванием около 13 лет. Синдром Гетчинсона-Гилфорда вызывает массовые изменения в организме больного, которые характерны для ускоренного и преждевременного старения. У больных в детском возрасте появляются классические старческие заболевания: проблемы с сердцем, суставами, ухудшается память, зрение, слух, кожа становится дряблой и морщинистой.

Причина данного состояния кроется в одной единственной мутации, которая находится в гене LMNA, который ответственен за кодирование белка ламина. Ламин белок, который входит в состав оболочки клеточного ядра. При его нехватке или неправильном строении ядро клетки становится хрупким и нестабильным. Клетки не могут нормально делиться и воспроизводить себя и постепенно умирают.
Из-за того, что заболевание не имеет специфических синдромов до 2-3 летнего возраста, его диагностика может быть затруднена. Первыми дают о себе знать прекращение роста ребенка, хрупкость костей и увеличенная форма головы с довольно маленьким носом.
Заболевание неизлечимо, единственный метод, который мог бы полностью избавить больных от него, это генная терапия. Но все разработки в этой области находятся в зачаточном состоянии. Пока врачам и пациентам доступно только симптоматическое лечение данного синдрома.
Существуют целые семьи в которых данным заболеванием болеет 2 и более ребенка. Одна из них индийская семья Ханов, в которой более сразу 3 ребенка.
Также может быть интересно: