Добрый день, уважаемые читатели. Сегодня довольно необычный выпуск, т.к. он предназначен в основном для людей с медицинским образованием и посвящен очень редкому заболеванию сердца - некомпактному миокарду.
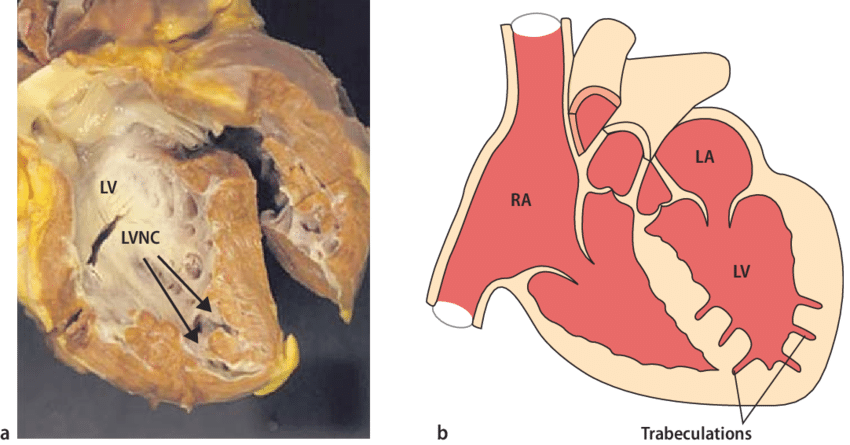
Некомпактная кардиомиопатия (НКМП, НМЛЖ) - это прогрессирующее заболевание, которое, как полагают, является следствием незавершенного развития миокарда. Характерными признаками заболевания являются повышенная трабекулярность миокарда (чаще левого желудочка) с наличием межтрабекулярных лакун, имеющих сообщение с полостью желудочка.
Основными осложнениями считаются нарушения ритма сердца (которые можно отслеживать портативно - СмартКардио) и тромбоэмболии. НКМП ассоциируется с повышенным риском внезапной смерти, особенно у лиц со сниженной фракцией выброса. Прогноз заболевания, как правило, неблагоприятный и существенно зависит от своевременной диагностики данной патологии. [3]
Впервые эта патология была описана S.Bellet и B.A. Gouley в 1932 году в сочетании с атрезией аортального клапана, затем была опубликована статья о постнатальном персистировании эмбрионального миокарда (1975) и описания губчатого миокарда у мужчины 62 лет, погибшего от внезапной сердечной смерти. Официальное признание некомпактный миокард получил после публикации работы Chin и коллег в 1990 году. В своей работе авторы не только описали патологию, но и предложили первые эхокардиографические критерии диагностики.
Причины заболевания
В настоящее время нет единого мнения относительно причины развития НКМП. Американская Ассоциация Сердца классифицирует некомпактный миокард как генетически детерменированную кардиомиопатию, связанную с задержкой нормального развития миокарда на 5-8 неделях внутриутробного развития. В противоположность этой позиции, Европейское Общество кардиологов (ESC) считает НКМП “неклассифицируемой кардиомиопатией”, которая может быть морфологическим проявлением различных видов кардиомиопатий.
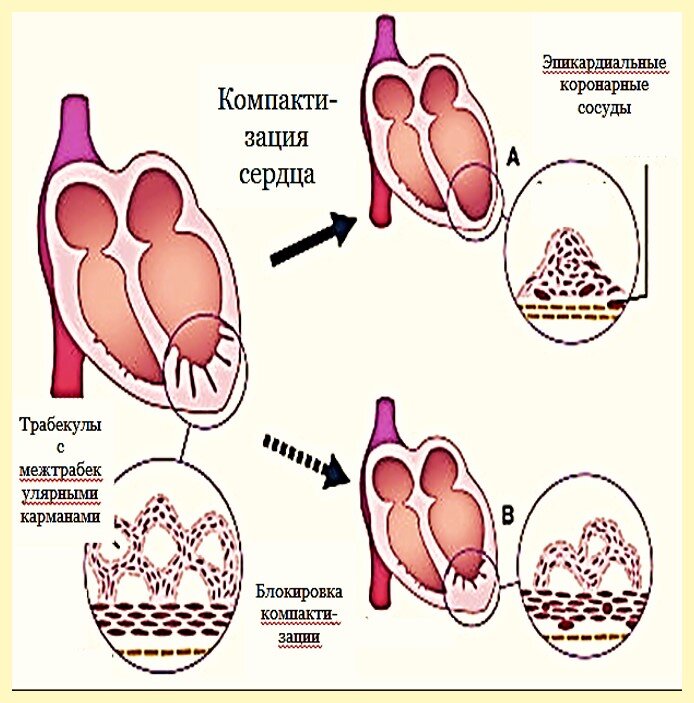
В поддержку генетической теории сказался тот факт, что были выявлены мутации в локусах, отвечающих за построение саркомеров (TMP1, MYH7, ACTC1, TNNT2, MYBPC3), Z-дисков, белка α – дистобревина, митохондриальные мутации, а также мутации в MIB1, RYR2 с более высокой частой встречающиеся у больных с НМЛЖ, чем в здоровой популяции. [2, 4, 5] Также имеются данные, доказывающие связь синусовой брадикардии, нередко выявляемой у больных НМЛЖ, с мутациями в гене, кодирующим ионные каналы - HCN4. [6] Выявление семейных форм заболевания служит подтверждением существования генетически детерменированных причин возникновения НМЛЖ (так же, как и при гипертрофической кардиомиопатии).
НМЛЖ в 12% случаев сочетается с врожденным пороком сердца, таким, как стеноз выводного тракта ЛЖ (46%, главным образом - двустворчатый аортальный клапан, читайте подробнее об этой патологии в статье), аномалия Эбштейна (25%), тетрада Фалло (8%), удвоение выводного отверстия правого желудочка (4%), миокардиальный мост (прохождение одной из магистральных коронарных артерий через миокард ЛЖ). Имеются сообщения об обнаружении некомпактности миокарда у больных с синдромами Шерешевского-Тернера (заболевание, связанное с моносомией по Х-хромосоме), Барта (патология, вызванная мутацией в гене, кодирующим белок таффазин). [7]
Ряд исследований подтверждают возможность сочетания НМЛЖ с нейро-мышечной дистрофией (НМД) (в том числе метаболической миопатией, наследственной оптической нейропатией Лебера, миотонической дистрофией, мышечной дистрофией Бекера, синдромом полимиелита, мышечной дистрофией Дюшена, MYH7 миопатией), что было показано как неблагоприятный прогностический фактор. [8]
Причиной возникновения теории развития НМЛЖ как результата ремоделирования сердца стал ряд исследований, подтверждающих формирование некомпактности миокарда в ответ на увеличение пред/постнагрузки на сердце. Наличие повышенной трабекулярности было продемонстрировано примерно в 30% случаев у больных с увеличенной преднагрузкой на ЛЖ (с почечной недостаточностью, поражением клапанов сердца, серповидноклеточной анемией), а также у спортсменов и беременных женщин. [9, 10]
Первым подтверждением данной теории стала работа Gati и коллег, в которой они исследовали 102 беременных женщины (не имевших признаков поражения сердечно-сосудистой системы) как естественную модель увеличенной преднагрузки на сердце. У 26 из них была зарегистрирована повышенная трабекулярность, развившаяся во время беременности и подвергнувшаяся обратному развитию через 2 года после родоразрешения.. В исследовании с участием 1000 клинически бессимптомных спортсменов, 18% из них имели признаки повышенной трабекулярности, а 8% подходили под критерии НМЛЖ, что ставило вопрос об адаптивном, либо прогрессирующем неблагоприятном характере морфологических изменений миокарда с вытекающим из этого требованием ограничения занятий спортом.
Распространенность заболевания
Статистические данные о распространенности НМЛЖ на Земном шаре варьируют от 3 до 9% от общего числа детей, страдающих кардиомиопатиями [9, 11].
Клиническая картина
Основным клиническим проявлением НМЛЖ считается прогрессирующее нарушение сократительной способности левого желудочка (ЛЖ) с неизбежным развитием сердечной недостаточности. Основные осложнения – нарушения ритма сердца и тромбоэмболии. Манифестировать заболевание может с клиники сердечной недостаточности, с нарушения ритма сердца или выявляется случайно при проведении Эхо-КГ или МРТ сердца по поводу другой патологии.
Имеются сообщения об обнаружении повышенной трабекулярности у людей без клинической симптоматики, особенно у спортсменов. В связи с потенциальным риском развития внезапной сердечной смерти, проводились многолетние наблюдения за людьми, находящимися в группе риска, в том числе для решения вопроса об ограничении занятий профессиональным спортом. Был обозначен ряд отличительных признаков при сравнении группы людей, занимающихся профессиональным спортом, с больными НМЛЖ при наличии одинаковых находок на Эхо-КГ и МРТ в двух группах. Так, в исследовании Gati и коллег было показано, что у людей, занимающихся профессиональным спортом, признаки наличия НМЛЖ нередко были случайной находкой при проведении Эхо-КГ. По результатам исследования Stacey и коллег некомпактность миокарда ассоциирована с наличием систолической и/или диастолической дисфункции сердца. [12] У больных с НМЛЖ в 66% случаев выявляют диаметр ЛЖ в диастолу более 64 мм, ФВ<45%, скорость наполнения ЛЖ <9 см/с. Отличительные признаки были замечены на ЭКГ: у спортсменов, как правило, наблюдают инверсию зубца Т в отведениях V1 - V3, в то время как у больных НМЛЖ инверсию зубца Т регистрируют в нижнелатеральных отведениях. Провокация эктопической тахикардии при выполнении нагрузочных тестов подразумевает наличие патологии, как и регистрация задержки контрастирования на МРТ. Рекомендуется выполнять скрининг родственников по первой линии на наличии мутаций, характерных для НМЛЖ.
Прогноз
Прогноз заболевания напрямую коррелирует со сроком дебюта болезни: чем раньше появляются признаки дисфункции желудочков сердца, тем более неблагоприятный прогноз. Существует несколько морфологических типов ремоделирования миокарда, при каждом из которых особенности диагностики, клиническая картина заболевания и прогноз будут отличны от остальных. Большинство авторов склонны выделять следующие группы в зависимости от морфофункционального состояния миокарда: НМЛЖ с сохранной функцией ЛЖ; НМЛЖ в сочетании с дилатацией ЛЖ; НМЛЖ в сочетании с изменением сократительной функции ЛЖ по рестриктивному типу; НМЛЖ в сочетании с гипертрофией миокарда; НМЛЖ в сочетании с неклассифицируемой кардиомиопатией. В исследовании Jefferies и коллег, в котором приняло участие 155 больных НМЛЖ, было показано, что прогноз заболевания снижается в ряду: изолированный НМЛЖ - НМЛЖ в сочетании с дилатацией ЛЖ - НМЛЖ в сочетании с гипертрофией миокарда - НМЛЖ в сочетании с неклассифицируемой кардиомиопатией. Прогноз оценивался, с опорой на данные о частоте внезапной смерти, необходимость трансплантации сердца и возникновение осложнений со стороны сердечно-сосудистой системы. [7]
При сравнении групп детей с диагнозами дилатационная кардиомиопатия (ДКМП) и НМЛЖ с ремоделированием сердца по дилатационному типу было показано, что продолжительность жизни в среднем одинакова в двух группах. Частота осложнений со стороны сердечно-сосудистой системы несколько выше у детей с ДКМП ( 47% больных с ДКМП и у 37% больных НМЛЖ, протекающим по дилатационному типу). Напротив, при сравнении групп детей с гипертрофической кардиомиопатией (ГКМП) и детей с НМЛЖ, сочетающимся с гипертрофией миокарда, обнаружено, что прогноз заболевания значительно хуже при наличии участков некомпактности миокарда по сравнению с изолированной ГКМП [7].
Благодарим за внимание! Следующий выпуск будем посвящен диагностике этого заболевания.
Подписывайтесь на ТГ-канал СмартКардио для врачей!
Автор статьи - Кульбачинская Екатерина, врач детский кардиолог.
Список литературы
1. Altenberger J, Hasenauer G, Granitz M, Stollberger C, Finsterer J. Disappearance of left ventricular hypertrabeculation/noncompaction and sudden death in a patient with Turner mosaic syndrome. Am J Cardiol. 2012;110(2):314-315. doi:10.1016/j.amjcard.2012.02.070.
2. Shen Y, Li X, Lu D, Xiao A, Li J. Myocardial Noncompaction Presenting With Myocardial Bridge: A Case Report. Medicine (Baltimore). 2015;94(36):e1425. doi:10.1097/MD.0000000000001425.
3. Caselli S, Attenhofer Jost CH, Jenni R, Pelliccia A. Left Ventricular Noncompaction Diagnosis and Management Relevant to Pre-participation Screening of Athletes. Am J Cardiol. 2015;116(5):801-808. doi:10.1016/j.amjcard.2015.05.055.
4 Ojala T, Nupponen I, Saloranta C, et al. Fetal left ventricular noncompaction cardiomyopathy and fatal outcome due to complete deficiency of mitochondrial trifunctional protein. Eur J Pediatr. 2015;174(12):1689-1692. doi:10.1007/s00431-015-2574-9.
5. Vermeer AMC, van Engelen K, Postma A V, et al. Ebstein anomaly associated with left ventricular noncompaction: an autosomal dominant condition that can be caused by mutations in MYH7. Am J Med Genet C Semin Med Genet. 2013;163C(3):178-184. doi:10.1002/ajmg.c.31365.
6. Milano A, Vermeer AMC, Lodder EM, et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol. 2014;64(8):745-756. doi:10.1016/j.jacc.2014.05.045.
7. Jefferies JL, Wilkinson JD, Sleeper LA, et al. Cardiomyopathy Phenotypes and Outcomes for Children With Left Ventricular Myocardial Noncompaction: Results From the Pediatric Cardiomyopathy Registry. J Card Fail. 2015;21(11):877-884. doi:10.1016/j.cardfail.2015.06.381.
8. Stollberger C, Blazek G, Gessner M, Bichler K, Wegner C, Finsterer J. Neuromuscular comorbidity, heart failure, and atrial fibrillation as prognostic factors in left ventricular hypertrabeculation/noncompaction. Herz. 2015;40(6):906-911. doi:10.1007/s00059-015-4310-7.
9. Cheng TO. Left ventricular noncompaction cardiomyopathy: three decades of progress. Int J Cardiol. 2014;174(2):227-229. doi:10.1016/j.ijcard.2014.03.208.
10. Gati S, Rajani R, Carr-White GS, Chambers JB. Adult left ventricular noncompaction: reappraisal of current diagnostic imaging modalities. JACC Cardiovasc Imaging. 2014;7(12):1266-1275. doi:10.1016/j.jcmg.2014.09.005.
11. Niemann M, Stork S, Weidemann F. Left ventricular noncompaction cardiomyopathy: an overdiagnosed disease. Circulation. 2012;126(16):e240-3. doi:10.1161/CIRCULATIONAHA.112.095059.
12. Stacey RB, Andersen MM, St Clair M, Hundley WG, Thohan V. Comparison of systolic and diastolic criteria for isolated LV noncompaction in CMR. JACC Cardiovasc Imaging. 2013;6(9):931-940. doi:10.1016/j.jcmg.2013.01.014.