В данной статье мне хотелось бы остановиться на таком важном вопросе как разработка воспроизведенного препарата, т.к. это важнейший этап для последующего доказательства биоэквивалентности. Нижеприведенная информация главным образом касается пероральных твердых лекарственных форм (таблеток, капсул) с немедленным высвобождением. Для лекарственных препаратов, представляющих собой пероральные или парентеральные растворы общие принципы описанные ниже также применимы, но в каждом случае имеются свои особенности.
1. Во-первых, воспроизведенный препарат должен быть в такой же лекарственной форме и максимально близок по качественному, и в идеале количественному составу к оригинальному/референтному препарату. Стоить помнить, что в рамках оценки биоэквивалентности различные пероральные лекарственные формы с немедленным высвобождением могут быть признаны одинаковыми. Например, таблетки и капсулы, диспергирующие лекформы, а также сиропы, пероральные растворы и суспензии могут сравниваться между собой, если они не всасываются во ротовой полости. Если оригинальный препарат представляет таблетки, а воспроизведенный капсулы или сироп, то ход фармацевтической разработки будет отличаться от описанного ниже.
В ходе фармацевтической разработки соответственно тщательно изучается оригинальный препарат (его состав, и его биофармацевтические свойства). На основе свойств лекарственного вещества, характеристик эталонного препарата и его информации в отношении, срока годности, упаковки, условий хранения, и т. д. определяется профиль качества целевого продукта (quality target product profile, QTPP), который включает растворение, однородность дозированных единиц, родственное вещество и другие аспекты качества и эквивалентности продукта.
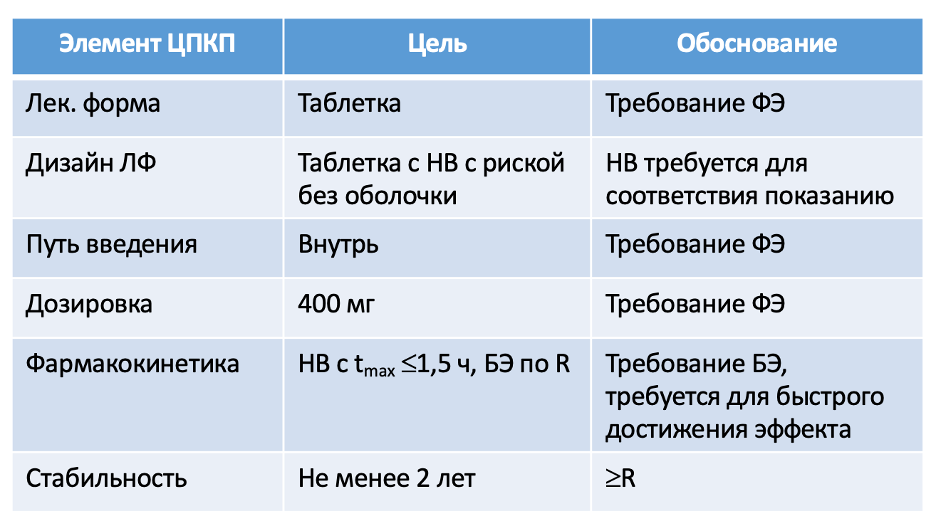
На основании этих данных и фармакопейных требований к лекарственной форме разрабатываются несколько формуляций воспроизведенного препарата (прототипы).
При этом выполняются различные испытания по оценке как действующего вещества [активного фармацевтического ингредиента, АФИ] (физико-химические характеристики вещества), которое будет использоваться при производстве воспроизведённого препарата, так и испытания его совместимости с вспомогательными веществами, которые планируются к использованию для формуляции препарата. Данные вспомогательные вещества не должны приводить к ухудшению показателей стабильности АФИ.
Также выполняются различные испытания нескольких прототипов формуляций препарата с различными комбинациями вспомогательных веществ для оптимизации их содержания. Оценивают такие показатели качества как распадаемость, растворение, стабильность и другие. После того как определен оптимальный прототип формуляции выполняют испытания для сравнения полученной формуляции с оригинальным препаратом.
3. Для целей оценки биоэквивалентности наиважнейшая оценка на данном этапе это растворение, так как для исследования биоэквивалентности нужно выбрать оптимальные серии как воспроизведенного препарата, так и референтного препарата. Т.е. сравнивают профиль высвобождения АФИ из нескольких серий исследуемого препарата в той дозировке в которой он будет изучаться в исследовании биоэквивалентности с аналогичной дозировкой референтного препарата, который также должен быть представлен в нескольких сериях (пункт 20 Правил исследований биоэквивалентности в ЕАЭС).
Соответственно в исследование биоэквивалентности должны отбираться те серии, которые показали наилучшие и максимально сопоставимые профили растворения. Данная серия воспроизведенного препарата называется «биосерией». Срок годности этой серии должен быть как можно большим, т.е. не следует изучать препарат со сроком годности истекающим в ближайшее время.
4. Содержание действующего вещества в биосерии не должно отличаться более чем на 5% от серии референтного препарата, т.е. например содержание периндоприла в R препарате 9,86 мг, содержание периндоприла в T препарате 9,88 мг, что соответствует требованию +/- 5%.
5. Также предъявляются особые требования к размеру серии (количеству единиц выпущенных лекарственных форм). Так пункт 22 Правил исследований биоэквивалентности в ЕАЭС гласит:
а) в отсутствие должных обоснований исследуемый лекарственный препарат должен быть отобран из серии, составляющей, по меньшей мере, 1/10 промышленной серии, или 100 000 единиц лекарственных форм, в зависимости от того, какой из объемов больше;
б) производство использованных серий лекарственного препарата должно обеспечивать высокую степень уверенности в том, что лекарственный препарат и процесс его производства будут воспроизведены в промышленном масштабе.
Объем серии, предназначенной для подтверждения биоэквивалентности, менее 100 000 единиц возможен при условии, что это предлагаемый объем серийного производства, и последующее масштабирование производственных серий не предполагается;
в) описание свойств и составление спецификации на такие критические показатели качества лекарственного препарата, как растворение, следует осуществлять, используя исследованную серию, т.е. серию, изученную в клинических исследованиях, в отношении которой подтверждена биоэквивалентность;
г) образцы лекарственного препарата из дополнительных опытно-промышленных и (или) промышленных серий, предоставленные на регистрацию, необходимо сравнить с образцами из серии, использованной в исследовании биоэквивалентности; они должны иметь сопоставимые профили растворения in vitro в подходящих условиях теста Растворение (согласно приложению N 5 к Правилам исследований биоэквивалентности в ЕАЭС).
И еще одно важное правило:
В отношении первых трех промышленных серий до выпуска их на рынок необходимо провести тест Растворение с биосерией и при несовпадении профилей растворения заявитель должен представить результаты испытания по собственной инициативе в уполномоченный орган и указать конкретные меры, предпринятые для преодоления возникшей ситуации.
Для прочих лекарственных форм с немедленным высвобождением системного действия, необходимо представить аналогичное подтверждение эквивалентности качества промышленных серий по отношению к исследованной серии.
Все это позволит показать то, что выпускаемые на фармацевтический рынок препараты с одной и той же производственной линии демонстрируют сопоставимость и подтверждают, что процесс производства будет выдавать продукт определенного качества, при неизменности всех условий производства. Другими научными словами это можно назвать валидацией процесса производства.
Соблюдение данных правил и норм позволит нашим пациентам получать приемлемые по качеству препараты. Споры про неэффективность воспроизведенных препаратов в данном случае не уместны, т.к. во всем Мире системы здравоохранения используют генерические препараты. Проблема генериков заключается в их качестве. Если в производстве используется качественная АФИ, и ее стабильность действительно сохраняется на всем протяжении срока годности, то препарат будет эффективен не менее чем на 80%, что доказывает исследование биоэквивалентности.
Как избежать ошибок при разработке препарата читай в этой статье и следи за ТГ каналом bioequivalence_in_EAEU.