Клеточный цикл
Клеточный цикл — это период существования клетки от момента ее образования путем деления материнской клетки до собственного деления или гибели. Какие-то из клеток — как, например, эпителий кишечника или стволовые клетки костного мозга — делятся на протяжении всей жизни, участвуя в регенерации тканей. Другие, напротив, достигнув высокого уровня специализации, делиться перестают. Таковы многие виды нейронов и клетки сердечной мышцы. Именно ролью отдельной клетки в организме в целом и определяется судьба каждой клетки — продолжать делиться; замереть, затормозив пролиферацию (сенесценция); или умереть, чтобы уступить место молодым (апоптоз). В основе любого из этих исходов лежат сложные биомолекулярные процессы, регулирующие разные аспекты жизнедеятельности клеток.
Рак нарушает порядок в этой слаженной системе регуляции. Клетки, «перешедшие на темную сторону», меняют траекторию развития, «забывая» нормальное предназначение и прежние функции в организме. Однако общие принципы регуляции клеточного цикла одинаковы и для нормальных клеток, и для опухолевых, с тем лишь отличием, что в последних либо выпадают отдельные элементы регуляции, либо, напротив, они чрезмерно активны.
В результате механизмы, необходимые для приостановки деления, в опухолевых клетках ослабевают; а процессы, в норме способствующие делению, напротив, усиливаются. Однако порой рак использует в своих интересах даже такие изначально анти-опухолевые процессы, как апоптоз.
В этой статье мы рассмотрим, как именно нарушения регуляции клеточного цикла в раке, а также апоптоз и сенесценция берутся на вооружение злокачественными опухолями; и как их изучают исследователи — видя в этих процессах потенциальные терапевтические мишени для борьбы с раком.
Логика клеточного цикла
Клеточный цикл — это процессы, происходящие от одного деления клетки до следующего. Приоритетная цель деления состоит в равномерном распределении неповрежденного генетического материала между двумя дочерними клетками. На обеспечение этого результата работают сложные молекулярные механизмы.
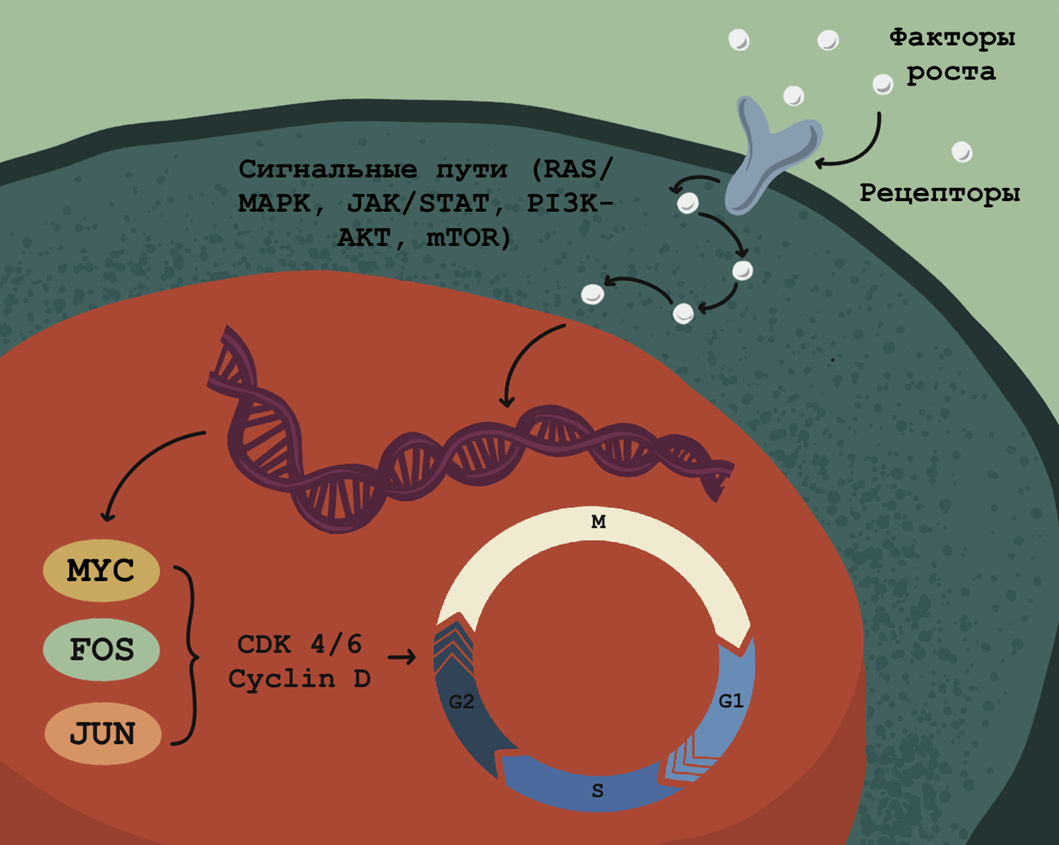
Начинается все с клеточных рецепторов, которые распознают особые молекулы — факторы роста (или митогены), подающие клетке сигнал к делению. Связывание ростовых факторов внеклеточными доменами этих трансмембранных рецепторов приводит к изменению структуры внутриклеточных доменов, которые в результате приобретают фосфорилирующую активность (способность модифицировать белки, присоединяя к ним остаток фосфорной кислоты).
Удивительно, но именно посредством простого присоединения или отрыва фосфорных групп от различных биомолекул, по сути, и обеспечивается вся сложность реакций клеточного цикла и протекание самого процесса клеточного деления. Подробнее о молекулярном устройстве ростовых рецепторов можно прочесть в статье, посвященной рецептору щелочи: «Рецептор „нетрадиционной ориентации“».
В клетке запускаются каскады реакций фосфорилирования одних белков другими. В конце концов, эта «молекулярная эстафета» достигает ядра, где комплекс циклин D-CDK4 фосфорилирует белок ретинобластомы (pRB), связанный с транскрипционным фактором E2F (pRB-E2F), что приводит к распаду этого комплекса. Высвобождение E2F запускает в ядре транскрипцию большого числа генов пролиферации и завершается физическим разделением клетки на две дочерние.
По подсчетам исследователей, в клеточном цикле задействовано более 750 различных белков (!) Такое множество молекул необходимо, чтобы обеспечить комплексную регуляцию процесса деления. Сложность сигнальных каскадов в делящейся клетке обеспечивает сразу несколько эффектов:
- активацию комплексов последующих стадий;
- инактивацию комплексов предыдущих стадий.
Эти условия способствуют однонаправленному протеканию деления, однако бесповоротность этого процесса в сочетании с основной его целью — распределением неповрежденной ДНК — рождает необходимость иметь «план Б». В том случае, если ее ДНК все-таки оказалась повреждена, клетка должна иметь возможность необратимо выйти из клеточного цикла. Такими запасными «путями отступления» для делящейся клетки являются апоптоз (запрограммированная клеточная смерть) и сенесценция (остановка клеточного цикла при сохранной метаболической активности).
Контрольные точки — клеточная проверка на дорогах
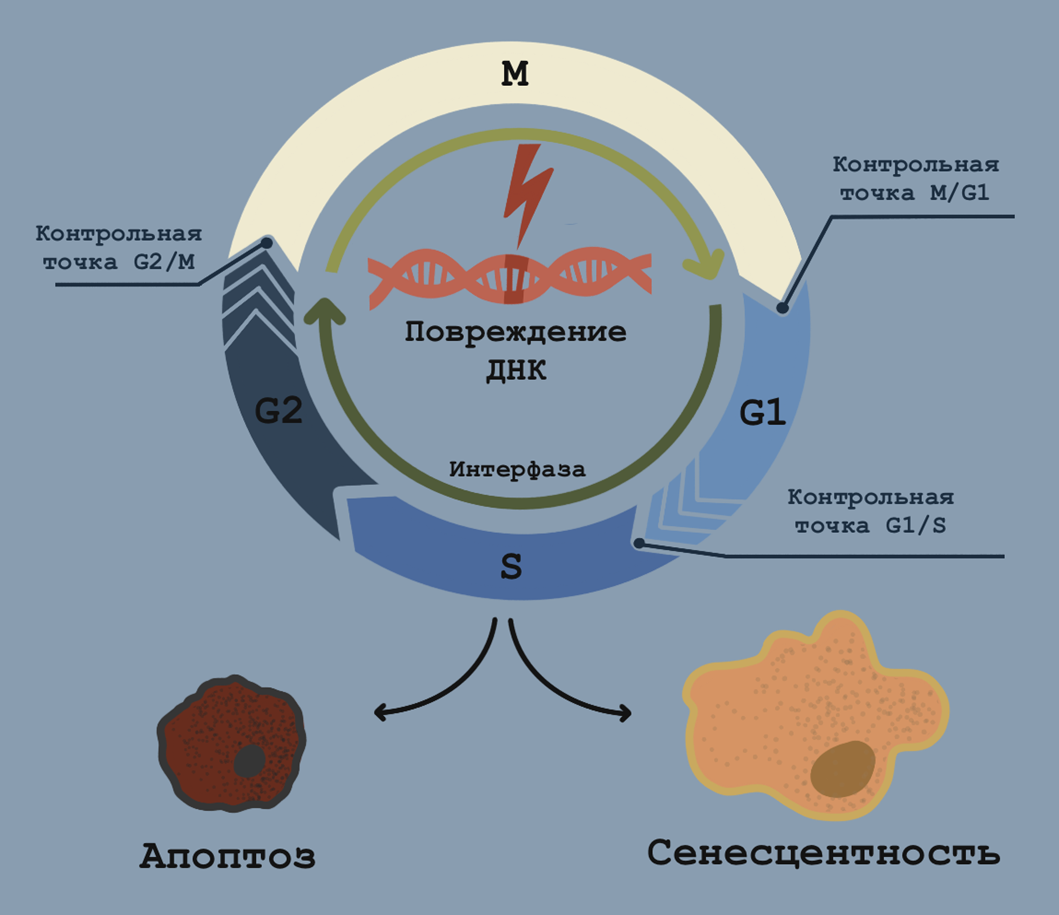
В клеточном цикле выделяют четыре основные фазы, проходя через которые, клетка претерпевает ряд значимых молекулярных событий. В пререпликативную G1-фазу происходит активный рост, синтез белков и АТФ, а также дупликация органелл. После чего в фазу S синтезируется копия ДНК (репликация), а в следующую фазу — G2 — клетка готова закончить подготовительный этап (интерфазу) и приступить собственно к митотическому делению (М-фазе). Последовательный и плавный переход из одной фазы в другую обеспечивается работой контрольных точек (чекпоинтов) — молекулярных регуляторов, распределенных по фазам клеточного цикла, в которых происходит «контроль качества» различных молекулярных событий. Всего таких чекпоинтов выделяют три и обозначают их по границе фаз клеточного цикла, переход между которыми и контролирует чекпоинт.
В течение всей интерфазы, пока деление еще не произошло, клетка убеждается, что ее ДНК не повреждена (G1/S-чекпоинт); в S-фазу — что структура репликативной вилки не нарушена и репликация прошла полностью (G2/M-чекпоинт); а в M-фазу — что веретено деления собралось правильным образом и каждая его нить несет отдельную хромосому (M/G1-чекпоинт). Выбор клеточной судьбы в результате работы чекпоинтов зависит от степени повреждения ДНК и фазы цикла, когда оно произошло. В пререпликативный период клетке доступны самые различные варианты, и, если она не подвергается апоптозу, то может уходить даже в обратимое состояние покоя (так называемый quiescence). На более поздних этапах интерфазы (S/G2), ближе к делению, играть с огнем мутаций становится опаснее, поэтому клетка прагматично выбирает один из сценариев выхода из цикла. Рассмотрим все три чекпоинта подробнее:
- G1/S: контрольная точка рестрикции. Здесь клетка принимает окончательное решение: двигаться ей дальше по клеточному циклу или нет, а для этого происходит проверка наличия мутаций в ДНК, не совместимых с продолжением деления. Поломки ДНК — вовсе не редкость (а, скорее, часть нормальной жизни): одни лишь двунитевые разрывы происходят по 10–50 раз в день в каждой клетке в случайные моменты интерфазы. Главное тут — каждое такое событие включает особую «сигнализацию», активирующую либо репарацию ДНК, либо замедление и остановку клеточного цикла.
Клеточная сигнализация: белки-сенсоры повреждения ДНК
Комплекс белков MRE1, RAD50 и NBS1 воспринимает сигнал повреждения ДНК и активирует киназу ATM (ataxia telangiectasia mutated). Главной мишенью ATM является белок CHK2, через который сигнал передается на «страж генома» — транскрипционный фактор p53; а затем на ингибитор циклин-зависимой киназы 2 (CDK2) — фактор p21. За счет блокирования CDK тормозится переход клетки из G1 в S-фазу. Вот так расшифровывается смысл обозначения G1/S.
- G2/M: контрольная точка интерфазы. Этот чекпоинт функционирует в течение S-фазы и отвечает на стресс, связанный с репликацией: от дисбаланса молекул, необходимых для репликации, до формирования трехмерных структур генома, создающих препятствия для прохождения репликативных комплексов. На этом этапе важную роль в протекании клеточного цикла играет киназа ATM, обеспечивающая как регуляцию раннего пререпликативного периода (G1), так и способная вызывать арест клеточного цикла в пострепликативный период (G2).
Трудности в копировании ДНК: что делать со сбойным конвейером
Скопировать целый геном — дело нелегкое и не лишенное технологических сбоев. Отсюда репликативный стресс, которому подвержены даже нормальные клетки в физиологических условиях. Чаще всего этот сбой выглядит так: ДНК-полимеразный комплекс, копирующий ген, слетает с него, оставляя свободный одноцепочечный конец ДНК. Накопление такой однонитевой ДНК в клетке последовательно активирует белки ATR и CHK1. Последний блокирует уже упомянутый белок CDC25 (Cell Division Cycle 25), что приводит к ингибированию циклин-зависимой киназы и к приостановке цикла. Во время данного чекпоинта клетке важно взять паузу, чтобы полностью закончить синтез ДНК и предотвратить повторную репликацию уже скопированных участков. Сформированные репликативные вилки в этот момент стабилизируются, ожидая «зеленого света» (= можно двигаться к митозу), однако их длительный простой заканчивается диссоциацией белков и последующим разрушением. В результате образуются все те же двуцепочечные разрывы ДНК, на которые клетка отвечает блокадой цикла и запуском каскада p53.
ATM — универсальный регулятор интерфазы
Комплекс белков MRE1, RAD50 и NBS1 воспринимает сигнал повреждения ДНК и активирует киназу ATM (ataxia telangiectasia mutated). Главной мишенью ATM является белок CHK2, через который сигнал передается на «страж генома» — транскрипционный фактор p53; а затем на ингибитор циклин-зависимой киназы 2 (CDK2) — фактор p21. За счет блокирования CDK тормозится переход клетки из G1 в S-фазу. Вот так расшифровывается смысл обозначения G1/S.
Киназа ATM проявляет свою активность преимущественно через фактор CHK2. В G2-фазе это приводит к деградации белка CDC25, роль которого — в тонкой настройке дефосфорилирования в клетке, соревнуясь с киназой WEE1, стремящейся, напротив, «повесить» на молекулы дополнительный ингибирующий остаток фосфорной кислоты. Заблокированный CDC25 уступает дорогу WEE1, и та выключает циклин-зависимую киназу 1 (CDK1), не давая наступить митозу — это и есть G2/M-блокирование.
- M/G1: контрольная точка сборки веретена. Для большей надежности существует и третий чекпоинт, работающий в M-фазу (проще говоря, во время митоза). Он отвечает на различные нарушения структуры веретена деления — в этот период тубулиновые микротрубочки начинают растаскивать хромосомы с экватора делящейся клетки к ее полюсам, чтобы обеспечить равномерное распределение ДНК между дочерними клетками. Если какая-то из хромосом не подцепилась или если обе сестринские хроматиды одновременно связались с одной нитью, возникает риск получить в результате деления клетки с неравным генетическим набором (кариотипом). Данную проблему и предотвращает чекпоинт M/G1 сборки веретена деления (spindle assembly checkpoint, SAC), восстанавливая генетический порядок.
- Инициация митоза — процесс необратимый, поэтому клетка не может выйти из него до тех пор, пока необходимость в чекпоинте сборки веретена не отпадает. Для клетки это означает одно — все хромосомы присоединены, порядок, едем дальше! Однако продолжительная активация SAC-комплекса (например, в случае терапии стабилизаторами микротрубочек — препаратами класса таксанов или алкалоидов винка) может завершиться уже знакомыми апоптозом или сенесценцией (либо так называемым «засыпанием» (mitotic slippage)). В этом случае митоз завершается без сегрегации хромосом и последующего цитокинеза, а клетка входит в новую интерфазу, будучи тетраплоидной.
SAC — контролер хромосомного порядка
Основным триггером на этом этапе выступает несвязанный кинетохор — белковый комплекс на хромосоме, в норме связанный с микротрубочкой. К такому одинокому кинетохору и привлекается мультибелковый комплекс SAC, который через каскад фосфорилирования угнетает активность фактора APC (anaphase promoting complex) — важного компонента продвижения по клеточному циклу. Интересно, что в экспериментах на клеточной линии PtK1, содержащей 12 хромосом, даже одного свободного кинетохора оказалось достаточно для стойкого ингибирования APC и ареста M-фазы. Таким образом, SAC замедляет митоз до тех пор, пока условие биполярного контакта с нитями веретена деления не будет достигнуто для всех хромосом без исключения.
Опухоль берет власть в свои руки. Чекпоинты и апоптоз
Логично предположить, что хаотичное деление опухолевых клеток запускается факторами, действующими непосредственно на пролиферацию. Однако результаты недавних исследований указывают, что неконтролируемое деление в раке в первую очередь вызвано мутациями в генах, в норме запускающих апоптоз в ответ на повреждение ДНК и способствующих выходу из клеточного цикла. То есть, не «зажать газ», а скорее «отключить тормоз». Патогенные мутации затрагивают все ключевые сигнальные белки как самого клеточного цикла, так и выхода из него. В этом смысле и перед самой опухолевой клеткой стоит парадоксальная задача — выключить чекпоинты, ограничивающие деление; при этом сохранить те, что обеспечивают генетическую сохранность (иначе не выжить и раковой клетке).
Интерфаза — единственный момент, когда клетка может выйти из цикла, если что-то пошло не так. Устранение клетки или ее обратимый выход в состояние покоя обеспечивается чекпоинтом повреждения ДНК (G1/S). Во многом этот выход зависит от активации p53-зависимого пути программированной клеточной гибели — апоптоза, запустить который могут как внешние, так и внутренние факторы (и в первую очередь это — повреждение ДНК). При апоптозе клеточная ДНК фрагментируется, и вся клетка распадается на аккуратно упакованные пузырьки — апоптотические тельца, которые затем поглощаются фагоцитами. Апоптозом управляет сложная сеть белок-белковых взаимодействий во главе с главными эффекторами — специальными протеазами каспазами.
Неудивительно, что мутации, инактивирующие p53, очень часто вызывают рак (подсчитано, что они встречаются в половине злокачественных новообразований). Однако, если мутации p53 препятствуют выходу из клеточного цикла (блокируя p53-индуцированный апоптоз), то существует и обратный механизм, позволяющий, напротив, неконтролируемо входить в него. Множество мутаций в протоонкогенах направлены как раз на эту часть биологии клетки (а в широком смысле — на поддержание активности транскрипционного фактора E2F, приоритетной мишени комплекса циклин D-CDK4/6). Таким образом, при раке нарушаются по меньшей мере два основополагающих механизма, останавливающих деление клеток с грубыми нарушениями ДНК.
При этом, если функционирование G1/S-чекпоинта в раке почти всегда и нарушено, два других чекпоинта — G2/M и M/G1, обеспечивающие сохранность ДНК активно делящихся опухолевых клеток, — продолжают в опухолевой клетке поддерживаться: это необходимо для выживания всех клеток, в том числе и раковых. Мутации генов, обеспечивающих работу данных чекпоинтов, встречаются в раке гораздо реже (по сравнению с тем же TP53).
По мере того, как в опухолевой клетке стимулируется деление, в ней усиливается инициация S-фазы (а значит, и последующее повреждение ДНК, обусловленное репликацией). В клетке возникает репликационный ДНК-стресс — индуцированный онкогенами. Данный вид стресса вызывает геномную нестабильность и, как считается, вносит вклад в формирование внутриопухолевой гетерогенности. Так как опухолевые клетки активно делятся, поддержание чекпоинта в S-фазе становится необходимым условием, чтобы избежать критического уровня повреждения ДНК при репликации, несовместимого с выживанием клетки. Логика здесь очень проста: клетка необратимо вошла в клеточный цикл, теперь ее план — выжить любой ценой.
На выполнение этого плана опухолевой клетки работает и M/G1-чекпоинт. Примечательно, что, несмотря на важность для развития рака, в небольшой доле опухолей наблюдается дефект данного чекпоинта: в частности, выявляются мутации белков, входящих в состав его основного компонента — комплекса сборки веретена деления (SAC). Опухолевые клетки во многом полагаются на стабильное функционирование этого комплекса, проводя в M-фазе в три раза больше времени, чем нормальные. Поэтому дисфункция SAC определяет возникновение одного из ярких признаков рака — анеуплоидии.
Ошибки при сегрегации хромосом в процессе деления, нарушения их количества и структуры порождают в опухолевых клетках хромосомную нестабильность (chromosome instability, CIN) — важное свойство некоторых опухолей, способное определять их клинический прогноз. С одной стороны, низкий уровень хромосомной нестабильности служит хорошим источником комбинативной изменчивости и способствует эволюции опухоли, участвуя даже в развитии резистентности к антиопухолевым препаратам. Высокий уровень CIN, напротив, вредит самой опухоли и ведет к стремительному накоплению летальных повреждений в ее клетках.
Однако интерпретация CIN high как прогностического маркёра вызывает больше дискуссий. В то время, как одни исследования указывают на улучшение прогнозов в некоторых локализациях и рака, другие выявляют снижение общей выживаемости пациентов с CIN high опухолями, что не позволяет использовать данный маркёр как универсальный.
Больше о рождении, старении и смерти клеток и о том, как знания об этом помогают лечить рак, читайте на нашем сайте!