Итак, пара сотен генов в геноме называются протоонкогенами. «Приобретая функцию», они становятся онкогенами, что неизбежно для многоклеточного организма, в котором обязательно есть системы регуляции роста тканей. Их действие в здоровых клетках уравновешивается активностью генов-супрессоров опухолей. Сигналы внутри клетки чаще всего можно свести к модели баланса между активирующими и ингибирующими — там нет одного «гонца», которого может перехватить один «страж»; скорее, речь будет идти о волне активирующих сигналов, которая сталкивается с волной ингибирующих сигналов, и тут уж кто кого переупрямит. Так, при обнаружении мутации в последовательности ДНК (может быть в протоонкогене) накопление сигнала, ведущего к аресту клеточного цикла, остановит переход в новую фазу. Но не всегда в мутации дело. Существует класс мощных протоонкогенов, способных изменять паттерны экспрессии генов в очень широком диапазоне, и они не требуют мутаций в кодирующей последовательности.
Факторы транскрипции (ФТ) —
очень могущественная группа генов, чьи продукты прямо или косвенно оказывают влияние на экспрессию сотен и даже тысяч генов в клетке. В сущности, всего четыре ФТ — Oct4, Sox2, Klf4 и c-Myc — способны откатить клетку назад в ее дифференцировке и сделать из какого-нибудь фибробласта индуцированную стволовую клетку, способную переквалифицироваться, например, в нейрон.
Именно за это открытие Синъя Яманака (эти четыре ФТ так и названы — факторы Яманака) и Джон Гердон получили в 2012 году Нобелевскую премию по физиологии и медицине. И один из этих факторов — c-Myc — входит в топ самых мощных онкогенов. Ему даже не нужна мутация — достаточно нарушенного баланса экспрессии. Вместе со «звездой» молекулярной онкологии p53 это — один из наиболее часто активируемых онкогенов среди человеческих злокачественных опухолей.
c-Myc — клеточный акселератор
Активация c-Myc имеет очень широкий спектр последствий в развитии опухоли — если считать отличительные признаки (Hallmarks of cancer), то это пролиферация, самообновление, выживание клеток, геномная нестабильность, нарушения метаболизма и инвазивность, ангиогенез и избегание иммунного ответа.
c-Myc активирует экспрессию:
- Ключевых регуляторов клеточного цикла — циклинов A2, D2, E1: они отвечают на сигналы, запускающие процесс деления клетки (если у мыши еще сломать mTOR- или MAPK-каскад, через девять дней появится опухоль размером с горошину);
- Фактора инициации трансляции EIF4E и ряда рибосомных белков — приводит к глобальному повышению уровня биосинтеза белка (если mTOR еще не пострадал, то это неплохая прелюдия к идеальному шторму);
- Ключевых ферментов метаболизма — енолазы и лактатдегидрогеназы А — ведут к изменению метаболизма в опухолевых клетках.
c-Myc снижает транскрипцию:
- Онкосупрессора p21, прямого партнера p53;
- Белка клеточной адгезии N-кадгерина — открывает метастатический потенциал.
При лимфоме Беркитта c-Myc в результате хромосомной транслокации оказывается под промотором гена тяжелой цепи иммуноглобулина. Экспрессирующийся ген не затронут, но в итоге — очень агрессивная опухоль.
Регуляторы клеточного цикла
Циклин-зависимые киназы (CDK) регулируют переход из одной фазы клеточного цикла в другую, и как с такими полномочиями не быть протоонкогенами? Касается это не только «классических» CDKs (CDK1, 2, 4, 6): есть целый ряд других CDK, которые могут играть онкогенную или онкосупрессивную роль. А некоторые (CDK8, 12, 19) — сразу обе.
О многоликой CDK8
Ее роль — транскрипционная (дез)активация, а главный партнер — циклин C. Для ее активации необходимы медиаторный комплекс (это связующее звено между факторами транскрипции и РНК-полимеразой II; CDK8 активируют Med12-Med13), и ее активность может быть стимулирована p21. В злокачественных опухолях CDK8 была определена как один из главных регуляторов перехода из G2- в M-фазу. В колоректальном раке ее описали как регулятор активности β-катенина, а сигнальный путь Wnt/β-catenin контролирует пролиферацию, миграцию и выживание клеток. В гематологических опухолях в силу своей функции транскрипционной (ин)активации CDK8 влияет на глобальные паттерны экспрессии генов, — от ингибирования киназы ждут терапевтического эффекта.
Рецепторы факторов роста
Просто так клетки организма не растут. Процесс этот регулируется сразу несколькими способами: в самом общем виде клетка должна получить сигнал о том, что надо расти, что есть за счет чего расти, и не должно быть сигнала о том, что расти некуда. «Надо расти» — это сигналы от факторов роста (их в человеческом организме много: EGF — эпидермальный фактор роста, FGF — фактор роста фибробластов, PDGF — тромбоцитарный фактор роста, и так далее). В большинстве случаев сигнал факторов роста принимают рецепторные тирозинкиназы. У этих трансмембранных рецепторов всего одна трансмембранная ɑ-спираль: сигнал внутрь клетки передается за счет димеризации рецепторов. При онкогенных мутациях для димеризации и последующего аутофосфорилирования никакие сигналы уже не нужны.
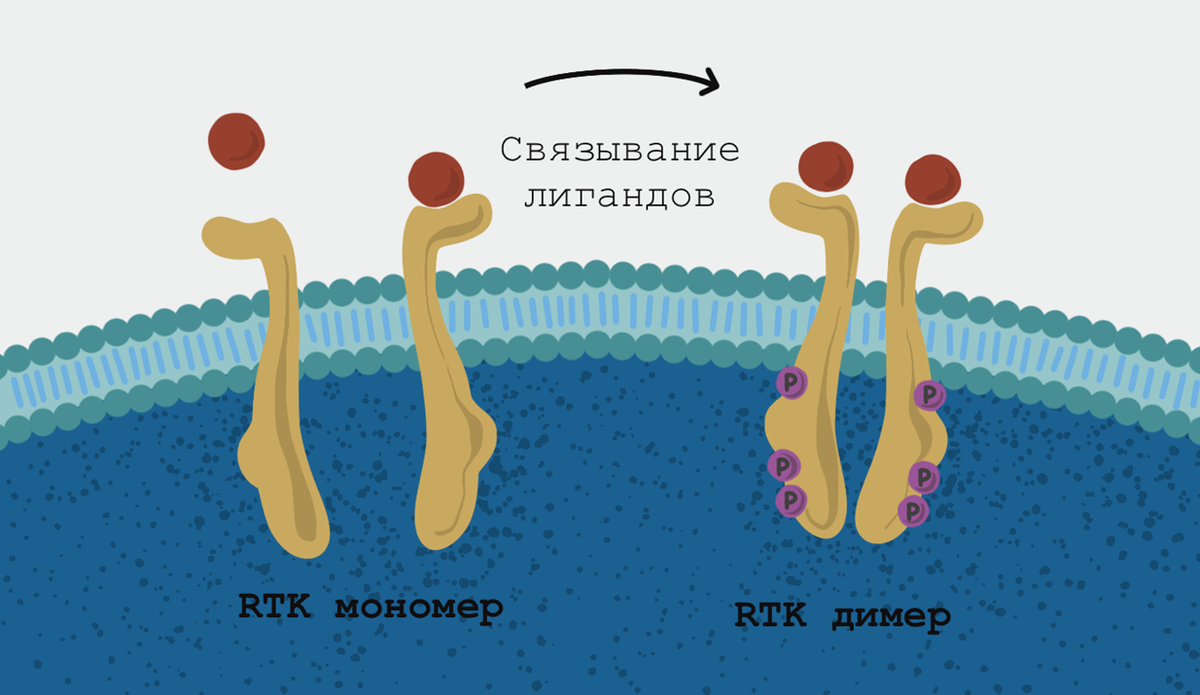
Рецептор эпидермального фактора роста — «антенна» для рака
Ниже по течению от EGFR (рецептор EGF) находятся сигнальные каскады mTOR, MAPK и STAT. Прекрасный набор, чтобы заставить клетку расти и делиться, что и происходит часто в немелкоклеточном раке легкого (NSCLC). Главным образом онкогенные мутации в EGFR — это мутации в киназном домене, прежде всего в 19 или 21 экзоне. Наиболее частая — замена лейцина на аргинин в 858 положении (L858R) в 21 экзоне (мутация 3 класса) или потеря от четырех до шести аминоксилот на участке от глутамата-746 до серина-752 (мутации 1 класса) — эти два класса составляют 85–90% всех встречающися в EGFR мутаций. По мутациям первого класса бьет ингибитор гефитиниб — он занимает место АТФ в АТФ-связывающем домене, и киназа не может фосфорилировать. И тут уж как повезет: нередко через 9–12 месяцев раковые клетки приобретают резистентность, или же опухоль окажется онкоген-зависимой — феномен, при котором рост и развитие опухоли опирается в основном на один онкоген, инактивация которого даже без химиотерапии приводит к уменьшению неоплазии (часто происходит с MYC-зависимыми опухолями, например).
G-белки
Семиспиральные GPCR (G-белоксопряженные рецепторы) после связывания с лигандом изменяют взаимную конформацию своих семи ɑ-спиралей, и это приводит к активации G-белка (бывают гетеротримерные и «малые»: и те, и другие имеют ГТФазную активность, необходимую для инактивации). Им помогают белки GEF и GAP: соответственно, заменяют «холостую» молекулу ГДФ на «боевую» ГТФ и позволяют отщипнуть от нее фосфат. Но G-белок — не киназа. Замена ГДФ на ГТФ осуществляется для активации G-белка: он меняет конформацию и может связаться и активировать Raf-белок. Если в результате мутации G-белок теряет ГТФазную активность, он остается в активной конформации.
KRAS — «замкнувший рубильник»
ГТФаза KRAS — один из наиболее часто мутирующих белков при раке поджелудочной железы (мутирует в почти 80% случаев): часто его мутации обнаруживают у пациентов с NSCLC, колоректальными опухолями. Самые частые мутации — замена глицина в 12 или 13 положении на аланин, цистеин, аспартат, валин. Хит-парад замен может различаться в зависимости от локализации опухоли: в раке поджелудочной G12D немного опережает G12V (39,2 и 32,5%), а в NSCLC — G12C (42%) вдвое опережает G12V. «Заставить замолчать» эти ГТФазы — мечта многих онкологов: «Ингибиторы Ras: в поисках Грааля таргетной терапии» и «Что мы узнали о биологии рака из клинических исследований. Несколько новелл об онкологии».
KRAS и его родственники (NRAS и HRAS) активируют MAPK каскад через Raf-белки, тоже известные онкогены. Впрочем, какой бы белок в этом каскаде не оказался мутантным, приобретя конститутивную активность, глобально эффект будет тот же. Но справедливости ради стоит сказать, что в пути RAS—RAF—MEK—ERK последние два эффектора мутируют значительно реже.
Безостановочные сигналы к пролиферации создают большую нагрузку на репликационную машину. Мутация ДНК-полимеразы приводит к лавинообразному накоплению мутаций. Например, в CpG-островках, которые часто бывают метилированы по цитозину, тот часто заменяется на тимин, очень похожий на 5-метилцитозин. Как бы то ни было, множественные нарушения целостности генома, а также стресс, который испытывает клетка с нарушенными сигнальными путями и метаболизмом, должны бы привести ее к апоптозу.
Но не тут-то было. Апоптоз, видимо, часто случается на ранних стадиях развития опухоли, как фактор отбора: выживают те, кто научился его избегать: активирующие мутации в антиапоптотическом гене Bcl-2 — весьма ценное приобретение для опухолевых клеток.
Онкосупрессоры
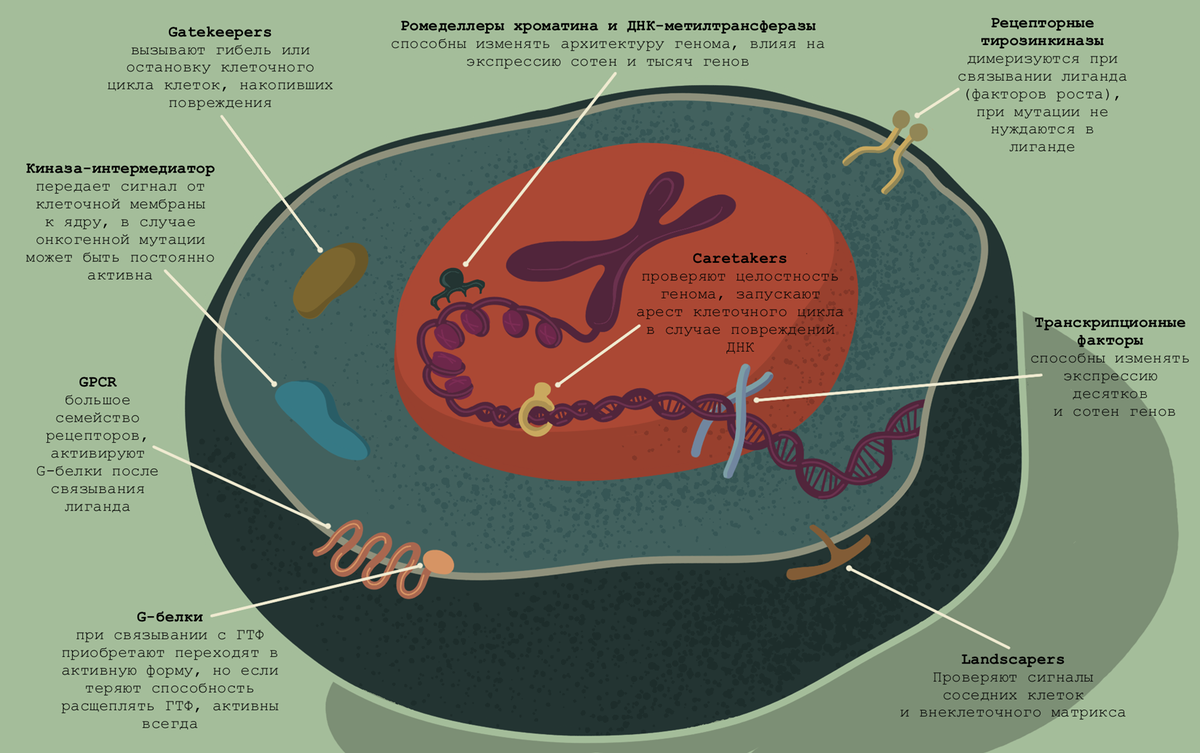
Онкогенные мутации обладают большим потенциалом, но было бы оскорбительно в отношении эволюции думать, что за долгие годы не нашлось, что противопоставить разрушительному потенциалу мутаций в протоонкогенах. В современной научной литературе выделяют три типа генов-онкосупрессоров. Разделение весьма условно. Gatekeepers — гены-привратники — контролируют клеточный цикл и проверяют, насколько правомерен переход к следующей фазе. Caretakers — смотрители, стражники генома; их задача — остановить процесс деления, если в геном вкралась ошибка, исправить ее, а если это невозможно — запустить апоптоз. Сравнительно недавно была сформулирована третья группа — landscapers. Они контролируют внеклеточный матрикс, следят за сигналами внеклеточной среды.
Gatekeepers — гены-«привратники»
Один из наиболее изученных генов-привратников — APC. Он впервые был описан в связи с семейными случаями колоректального рака, но впоследствии стало ясно, что и спорадическое развитие опухоли тоже часто происходит из-за того, что он не выполняет свои многочисленные функции. Одна из них — разрушение β-катенина (белка, регулирующего факторы транскрипции). Часто гены экспрессируются для того, чтобы их продукт был тут же отправлен на деградацию. Когда в нужный момент придет сигнал, этот процесс будет остановлен, и эффект будет почти мгновенный. Помимо APC, к принудительной конститутивной деградации β-катенина причастны еще AXIN, GSK-3, CK-1 и β-trcp. Но они не подстраховывают друг друга — наоборот, как в кружке революционера Нечаева — «повязаны кровью». Даст слабину один — и вся затея провалится. Так что и AXIN, и GSK-3 — тоже очень известные привратники.
А Wnt/β-catenin — канонический — сигнальный путь играет важную роль в пролиферации, а неакононический — в поляризации и миграции. Здесь вам и развитие опухоли, и метастазирование.
Другой классический привратник — SMAD4 — играет ключевую роль в TGF-β сигнальном пути. Он может заблокировать переход клеток из G1- в S-фазу и стимулировать транскрипцию p21.
Забавный факт, показывающий, что это многофигурная, но всё же композиция, все действующие лица друг с другом связаны: SMAD4 может ингибировать индуцированный эпидермальным фактором роста (EGF) сигнал, идущий по пути Wnt/β-catenin.
Caretakers — «стражи генома»
Гены MSH2 и MLH1 участвуют в репарации мисматчей — ошибок, при которых неправильно подбирается пара нуклеотиду матричной ДНК. Мутации в этих генах (а еще в MSH6, PMS2 и EPCAM) вызывают так называемый синдром Линча — наследственную предрасположенность и высокий риск развития в молодом возрасте колоректального рака или карциномы эндометрия. Кстати, если вы еще не забыли вступление к этой статье, семейные случаи рака, тем более его раннего развития — это повод нанести визит медицинскому генетику.
На двухцепочечные разрывы ДНК реагирует протеинкиназа ATM. Одна из ее мишеней — p53: важный онкосупрессор, речь о котором впереди. Если разрывы ДНК многочисленны, то через ATM запускается индукция апоптоза или сенесценция клетки.
Опухолевые супрессоры даже формируют раковую повестку в медиапространстве именно в связи с наследственным опухолевым синдромом, от которого никто не застрахован: даже если вы богаты, красивы и знамениты, мутация в BRCA1 может заставить вас пойти крайние меры. Кроме его паралога BRCA2, можно вспомнить такие важные и хорошо изученные онкосупрессоры, как RB1, CHK1/2 и PTEN. Про каждый из них можно написать немало, но перейдем, пожалуй, к неоспоримой звезде молекулярной онкологии.
Знаменитый p53 — один из самых изученных белков. Это и неудивительно, учитывая, что мутации в его гене встречаются более чем в половине всех опухолей. Особенность этого онкосупрессора в том, что он является исключением из двухударной модели Кнудсона: чтобы купировать его функциональность и дать дорогу накоплению мутаций, достаточно мутации в одном аллеле. Дело в том, что активная форма белка — тетрамер из четырех p53. И если один из аллелей несет мутацию (чаще всего в ДНК-связывающем домене), то сформировать функциональную единицу шансов гораздо меньше.
Количество функций p53, помимо стражи генома, огромно — он важен для пролиферации и апоптоза, задействован в аутофагии, дифференцировке и сенесценции клеток, ассоциирован с фертильностью и продолжительностью жизни, важен для приспособления к внешней среде и еще много для чего. Невольно задаешься вопросом: только p53 такой важный, или мы знаем о нем так много просто потому, что его исследовали вдоль и поперек?
p53 важен, но его накопление токсично для клеток, поэтому он находится под строгим контролем белка MDM2, который при каждой встрече отправляет p53 в протеасому для деградации. В свою очередь, p53 (он же фактор транскрипции!) активирует транскрипцию MDM2. Вот такая петля обратной связи. Чтобы нарушить связывание с MDM2 и последующую деградацию, p53 нужно фосфорилирование на N-концевом домене, а для этого нужно собраться в тетрамер. Значит, если тетрамеризация вследствие мутации невозможна, p53 не в состоянии и связаться с ДНК, и накопиться в клетке, чтобы запустить арест клеточного цикла, системы репарации или апоптоз. Не понятно одно — он всё-таки gatekeeper или caretaker?
Больше о генетических (и эпигенетических) причинах развития раковых опухолей, читайте в нашей новой статье!