В прошлый раз я рассказала, как работать с контигами в Geneious. Допустим, вы их просмотрели, приняли решение по каждому нуклеотиду. Обрезали, если посчитали нужным. Что дальше?
Для анализа надо сделать выравнивание. Оно нужно для того, чтобы сопоставить позиции разных последовательностей друг с другом, и понять, в какой позиции произошли замены.
Ниже я привожу ссылки на некоторые видео о том, как работают разные алгоритмы. Однако таких видео в интернете много, они разные по длительности, по освещаемым аспектам, на разных языках.
Чтобы детально понимать алгоритмы, надо углубиться в математику. Но на самом деле, на базовых уровнях работы с филогенетикой, слишком детальное понимание не особо нужно. Если вы будете заниматься биоинформатикой и решите сами писать скрипты, то там уже придется всю эту кухню осознать. Для начала работы с выравниваниями надо понимать следующие вещи:
1) Выравнивания бывают локальные и глобальные.
Глобальное выравнивание используется, когда вы работаете с последовательностями, сопоставимыми по размеру. Например, у вам несколько последовательностей одного и того же генетического маркера, но из разных организмов.
Локальное выравнивание нужно, когда вы хотите картировать маркер на более длинный участок ДНК. Например, у вас есть последовательность, и вы хотите узнать, какому участку генома она соответствует Тогда вам нужно взять аннотированный геном этого же вида или близкого. Аннотированный геном означает, что в файле, где он записан, промаркированы начало и конец генов и других участков генома. Даже если вы работаете с обычным баркод (цитохром оксидаза с субъединица 1 или COI, или cox1), иногда важно узнать, с какого конкретно нуклеотида по порядку в митохондриальном геноме начинается анализируемый вами ген. Например, это нужно, если вы хотите включить замены нуклеотидов в диагноз описываемого вами вида, и вам надо предоставить конкретную позицию замены в митохнодриальном геноме. Кстати, BLAST - это тоже локальное выравнивание. О нем я напишу отдельно чуть позже.
2) Разные алгоритмы выравниваний отличаются по точности и времени построения выравниваний. Вам нужно подобрать алгоритм в зависимости от вашего конкретного случая. Самые популярные - это ClustalW, Muscle и T-Coffee.
3) Какой бы алгоритм вы не использовали, результат выравнивания следует просмотреть и убедиться, что вас оно устраивает. Если какая-то последовательность выравнена некорректно, это повлияет на все последующие анализы. Такое может случиться по разным причинам. Например, она прочитана не с прямого, а с обратного праймера. В последовательности может быть контаминация, из-за чего в ней появились длинные вставки. Или наоборот, из-за плохого качества в сиквенсе много пропусков. Все подозрительные места надо просмотреть, и решить, что делать в каждом из случаев.
Мы остановились на редактировании контигов, и выглядят они вот так.
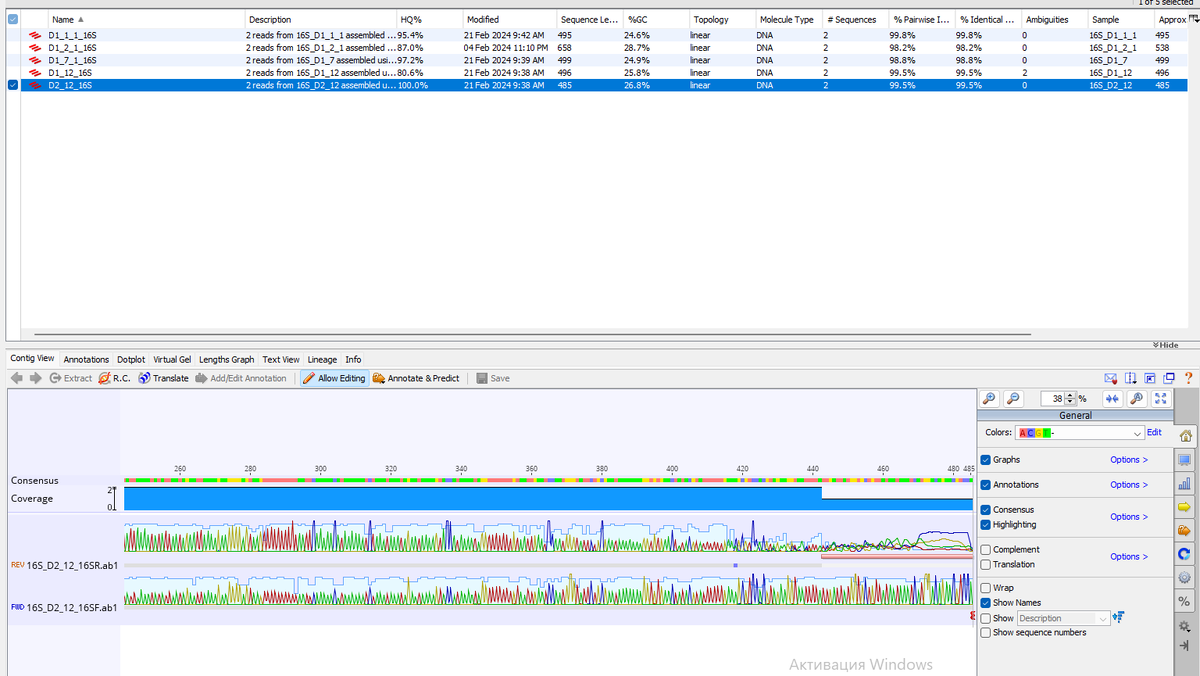
Давайте сделаем из них выравнивание.
Надо выделить все последовательности, и нажать на правую кнопку мыши. В выпадающем меню выбрать Multiple Align.
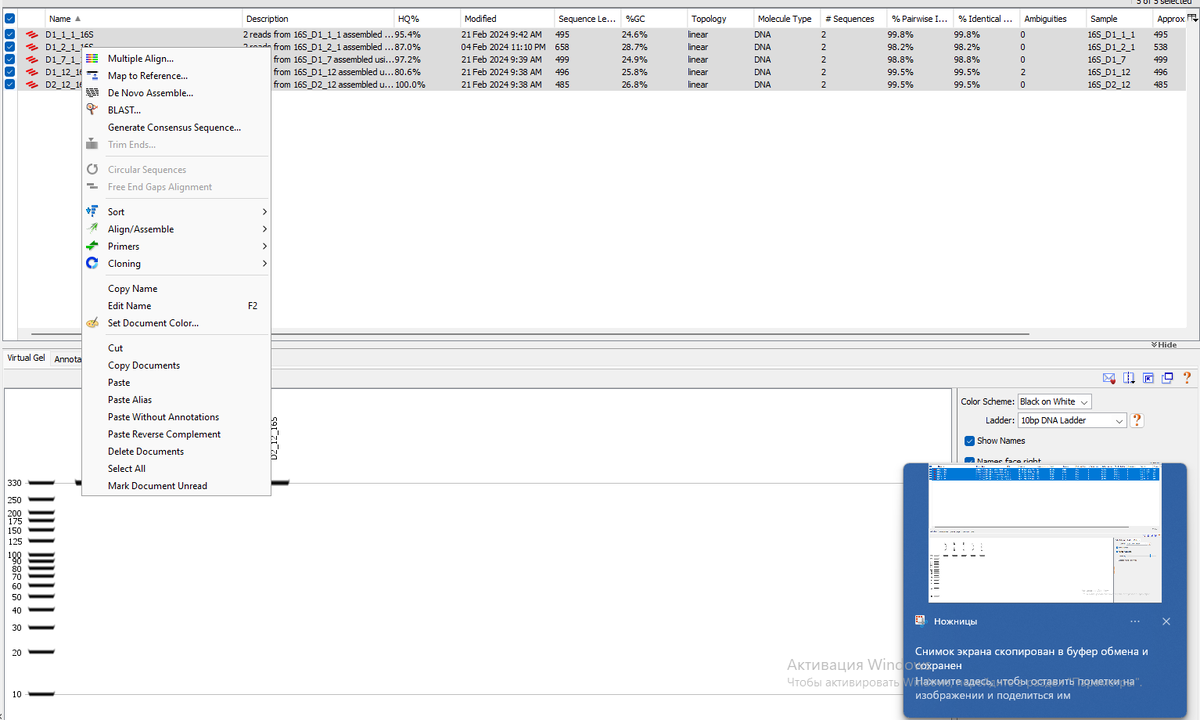
Появится вот такое окно. В данном случае нас интересует глобальное выравнивание. Также можно выбрать строгость выравнивания, по умолчанию стоит 65%. Можно так и оставить.
Как видно в данном случае программа не позволяет нам выбрать алгоритм выравнивания, потому что мы выравниваем консенсусы.
После того, как нажали на ОК, в списке появляется еще один файл, который называется Nucleotide Alignment. И внизу появляется выравнивание.
Поскольку мы выравнивали контиги, то тут их видим. По сути программа выровняла прочтения с обоих праймеров. Но это не совсем то, что нам нужно, потому что мы все-таки будем работать с выравниваниями консенусных последовательностей.
По сути можно сделать одну последовательность из контига, и все места, в которых была неопределенность будут обозначены соответственными буквами, такая последовательность и будет называться "консенсусной". В чем суть? Например, если в прочтении в одной и той же позиции по первому праймеру проставлен аденин (А), а в прочтении по второму праймеру проставлен тимин (Т), то в этой позиции будет стоять буква W в консенсусе. Другой вариант, если в двух прочтениях в одной позиции буквы отличаются, но в одной из них пик грязный, то в консенсусе подставится та буква, где пик чистый. Если у вас качественное прочтение с обоих праймеров и нет гетерозиготности, то во всех позициях по обоим прочтениям должны быть идентичные нуклеотиды.
Например, вот у нас последовательность D1_12_16S
В позиции 411 прочтения различаются. В одном С, в другом G. Оба пика грязные, но в каждом случае разные нуклеотиды несут более сильный сигнал. В контигах консенсус показан сверху цветными буквами, для этой позиции у него проставлен S (что значит, либо C, либо G).
Чтобы вывести консенсус в отдельный файл, надо выделить нужный контиг, нажать на него правой кнопкой мыши, и потом нажать на Generate Consensus Sequence.
Откроется вот такое окно. Можно оставить настройки по умолчанию.
Нажимаем ОК, Консенсус появляется отдельным файлом в списке.
Как видно, в позиции 411 буква S.
Вот еще один пример. Контиг D1_2_1_16S.
Тут в некоторых позициях есть различия в прочтениях. Но нижнее прочтение более грязное, поэтому программа подставляет значения более чистых пиков в консенсус. Однако если вы не согласны, то можете исправить вручную.
Следующая задача - сделать из всех контигов отдельные файлы с консенсусными последовательностями, также, как описано выше. И лучше всего положить их в отдельную папку. Как создавать папки см. здесь. Перенести последовательности можно элементарно с помощью Copy-Paste (CtrlC-CtrlV).
Вот как это выглядит у меня. Внизу показаны все консенсусные последовательности, но они еще не выровнены. Также нужно нажать на Multiple Align.
При работе с консенсусами появляется больше опций выравниваний.
Я выравниванию обычно с помощью Geneious Alignment. Но можно выбрать любой алгоритм, и посмотреть, чем они будут в реальности отличаться.
Ну вот, выравнивание сделано. И оно помещено в отдельный файл в списке под названием Nucleotide Alignment.