Я приступаю к следующему блоку своего блога - это обработка последовательностей и их анализ.
Прежде чем перейти к конкретным шагам, я хочу сделать небольшой дисклеймер о факторах, которые надо учитывать. Дело в том, что работа с последовательностями очень зависит от ваших целей и задач. Они влияют на то, как лучше работать с данными. Второй немаловажный фактор - это опыт конкретного человека, который вас учит. Я самоучка, никогда не проходила курса по молекулярной филогенетике, и училась у разных людей, которые не ставили целью дать мне какие-то систематические знания или четко объяснить логические цепочки. Я сама выбирала, что учить, а что нет, что читать, а что нет. И большинство людей, которые занимаются молекулярной филогенетикой, которых я знаю, учились примерно так же, и все мы различаемся в наших знаниях и предпочтениях. Третий фактор, заключается в том, что методы, связанные с последовательностями ДНК постоянно развиваются и уследить за ними довольно непросто. Наверняка, есть более эффективные способы делать что-то из того, что я делаю я, просто я пока о них не знаю. В связи с этим рекомендую вам изучить разные подходы к одним и тем же анализам, и решить для себя, что вам подходит лучше. Ну и, надеюсь, то, что напишу я, вам тоже будет полезным.
На мой взгляд, самая лучшая программа для обработки сырых последовательностей - это Geneious https://www.geneious.com/ К сожалению, она платная и довольно дорогая. Но если у вас есть возможность приобрести эту программу, то я вам ее рекомендую. У нее также есть бесплатный пробный период, и можно воспользоваться им на какое-то время.
На секвенирование по Сэнгеру вы отправляете ваши почищенные (а иногда не почищенные) пробы и праймеры. Праймеры обычно те, с которыми вы ставите ПЦР.
Файлы, которые вам присылают с секвенирования по Сэнгеру, имеют разрешение .ab1 Их обычно присылают в архиве, и когда вы его распаковываете, то файлы выглядят примерно вот так:
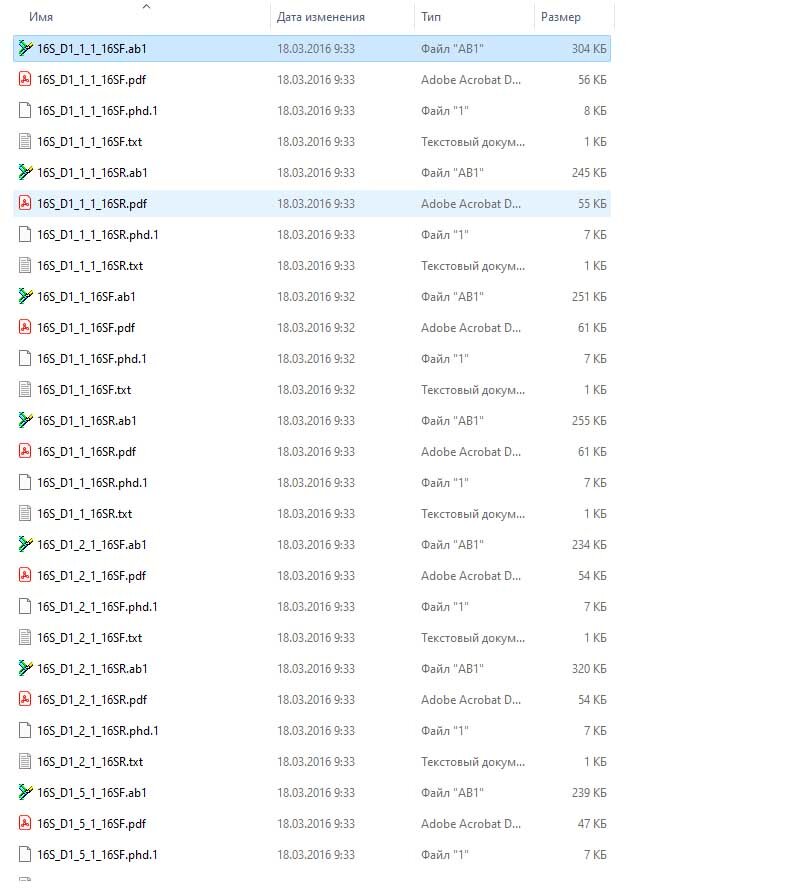
Тут еще есть и вспомогательные файлы, но загружать и анализировать вы будете только .ab1 Первая часть названия файла, например, 16S_D1_1_1, - это название вашей пробы. По сути то, что написано на той пробирке, которую вы отправляете на секвенирование. Вторая часть, например, 16SF или 16SR, - это праймеры, которые вы с ними отправляете. Я отправляю с двумя, прямым (F) и обратным (R), праймерами. Поэтому у меня для каждой пробы два сиквенса. Кто-то может отправить только с одним праймером, тогда у него для каждой пробы будет один сиквенс. Ну и как, наверное, понятно, в данном случае я отправляла на секвенирование митохондриальный маркер 16S rRNA.
Вот так выглядит Geneious, когда вы его открываете. Сначала может показаться, что программа не очень понятная, но на самом деле она довольно интуитивная.
Слева идет список папок. Создать папку очень легко, просто нажав на одну из уже имеющихся папок правой кнопкой мыши.
Если нажать на кнопку New Folder, то программа создаст подпапку в той папке, на которую вы нажали.
Таким образом, я создала папку test специально для этого руководства, а в ней еще три подпапки - raw_sequences, contigs и сonsensus. Это то, как я обычно разделяю рабочие последовательности. Но вы можете разработать свой вариант, который вам удобен. Как видите, около каждой папки стоит 0, это значит, что в ней не лежит ни одной последовательности.
Чтобы туда что-то загрузить, надо нажать на папку, в которую вы хотите поместить последовательности. Сейчас я выбираю raw_sequences.
Далее, в главном меню надо выбрать File - Import - From File
Ну и далее надо найти те файлы, которые вы хотите обрабатывать в той папке на компьютере, куда вы их сохранили.
После того, как вы нажмете на кнопку "Import", файлы появятся в папке Geneious в которую вы их загружали, в данном случае в raw_sequences.
Около этой папки изменилось число, теперь оно 24. Это означает что в ней 24 последовательности.
Если нажать на любой из этих файлов, то появится соответствующая сырая последовательность.
Загрузка завершена.