Исследования биоэквивалентности направлены на сравнение двух лекарственных препаратов. Сравнение, может быть, по фармакокинетическим конечным точкам, фармакодинамическим конечным точкам или по параметрам клинической эффективности и безопасности. Согласно требованиям документов Евразийского экономического союза (ЕАЭС) принято, что исследования с оценкой фармакодинамики проводятся в случае невозможности или нецелесообразности выполнения фармакокинетических исследований, а исследования клинической эффективности/безопасности выполняются при невозможности или нецелесообразности выполнения фармакокинетических и фармакодинамических исследований.
Примером невозможности или нецелесообразности фармакокинетических исследований являются, например, препараты в лекарственных формах для парентерального введения, т.к. их биодоступность не ограничивается всасыванием в желудочно-кишечном тракте и поэтому они либо сразу, либо очень быстро попадают в системное кровообращение. Сравнивать их фармакокинетику нецелесообразно.
Для всех пероральных лекарственных препаратов, для доказательства биоэквивалентности в первую очередь следует рассматривать исследования, направленные на сравнение фармакокинетики.
Для этого нужно спланировать исследование, которое будет учитывать особенности фармакокинетики действующего вещества и особенности лекарственных форм сравниваемых препаратов.
В самом простом стандартном варианте исследования биоэквивалентности планируются как одноцентровые, двухпериодные, перекрестные исследования у здоровых добровольцев с однократным приемом натощак. В таких исследованиях каждый участник получает сначала один препарат, а затем, через период времени достаточный для полного выведения лекарства из крови (период отмывки), получает второй препарат. То есть участник исследования принимает оба сравниваемых препарата. Однако в зависимости от различных факторов дизайн и схема исследования могут значительно различаться.
Следует учитывать следующие особенности:

Безопасности и токсичность - определяют возможность участия в исследовании здоровых добровольцев или пациентов. Как правило у пациентов изучаются цитотоксические препараты, применяемые в онкологии, т.к. исследовать их у здоровых добровольцев не этично, и может быть опасным. В остальных случаях исследования проводятся у здоровых добровольцев, даже в случае препаратов с узким терапевтическим диапазоном. Также для таких не безопасных препаратов рекомендуется использовать альтернативный дизайн исследования – однопериодный в параллельных группах, чтобы каждый пациент получал только одну дозу лекарства. Соответственно такие исследования не требуют периода отмывки и по длительности короче исследований с перекрестным дизайном. Если препараты не безопасны для женщин (риск репродуктивной токсичности), то в исследовании должны принимать участие только субъекты мужского пола, также если препарат будет применяться только у женщин, то в исследование должны включаться только субъекты женского пола.
Период полувыведения – параметр, который определяет период отмывки (должен превышать 6 периодов полувыведения [t1/2]). Для препаратов с длительным периодом полувыведения (к таковым могут отнесены препараты с t1/2 более 24 часов) значительно увеличивается период отмывки, требуемый для полного выведения действующего вещества, что очень сильно затягивает проведение исследования во времени. Поэтому для таких препаратов следует рассмотреть альтернативный вариант дизайна исследования – параллельный, что сокращает длительность исследования. Также период полувыведения определяет достаточную длительность забора образцов крови для оценки фармакокинетического профиля сравниваемых препаратов (общая длительность забора крови должна превышать 4 t1/2).
Внутрииндивидуальная/межиндивидуальная вариабельность – это ключевые параметры для определения размера выборки в исследованиях биоэквивалентности с перекрестным и параллельным дизайном. Так для расчета размера выборки по данным внутрииндивидуальной вариабельности ранее выполненного исследования с перекрёстным дизайном, возможно планировать выборку только для исследования с перекрестным дизайном, и нельзя планировать размер выборки исследования с параллельным дизайном.
В то же время, если известны и внутрииндивидуальная и межиндивидуальная вариабельность, возможно рассчитать общую вариабельность и уже на основании этих данных оценивать размер выборки в исследования с параллельным дизайном.
В целом факторы, влияющие на вариабельность в исследованиях биоэквивалентности, можно разделить на контролируемые и неконтролируемые.
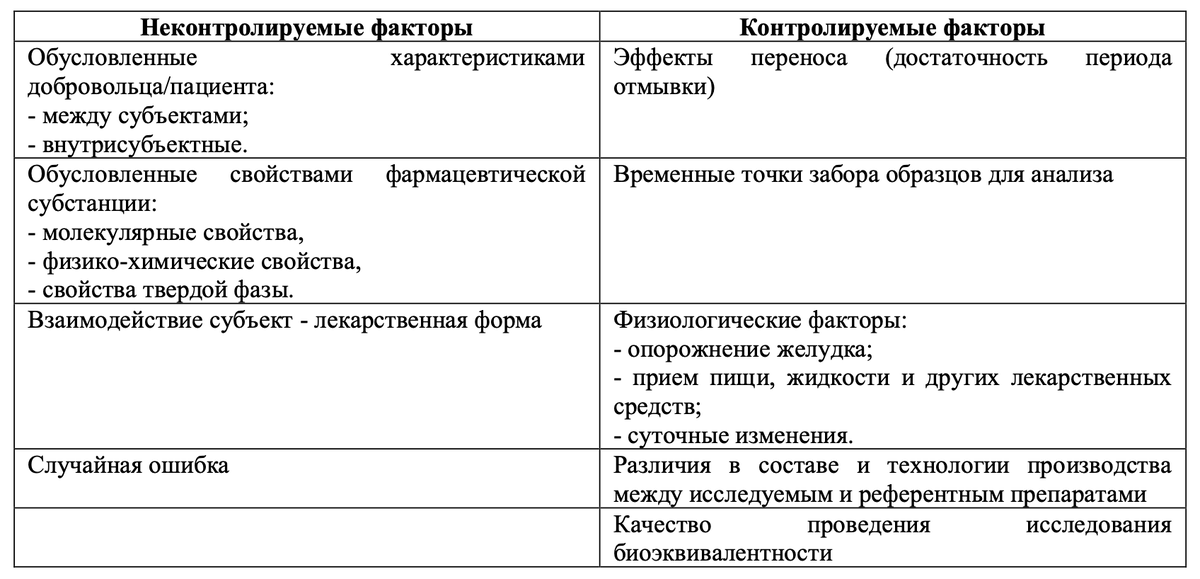
Влияние контролируемых факторов можно исключить надлежащим проведением исследования биоэквивалентности или надлежащей разработкой готового лекарственного препарата. Исключить влияние неконтролируемых факторов не представляется возможным, именно они и являются причиной отнесения лекарственных препаратов к высоковариабельным.
Влияние неконтролируемых факторов можно оценить, но только в исследованиях с перекрестным дизайном, т.к. статистическая оценка в таких исследованиях позволяет оценить вклад всех известных факторов влияющих на общую вариабельность и вычислить остаточную вариацию (остаточную ошибку), которая и является внутрисубъектной (внутрииндивидуальной) вариабельностью.
По результатам исследований с параллельным дизайном невозможно получить данные о внутрииндивидуальной и межиндивидуальной вариабельности, т.к. эти данные характерны только перекрестным исследованиям. Соответственно опираясь на данные исследований с параллельным дизайном, можно планировать размер выборки только для исследований с параллельным дизайном.
Условия приема натощак или с пищей – если референтный (оригинальный) препарат применяется натощак, то исследование проводиться натощак, если с приемом пищи, то и исследование проводится в соответствующих условиях. Однако для препаратов в лекарственных формах с модифицированным высвобождением, требуется изучение при обоих состояниях.
Время достижения максимальной концентрации (Cmax) – определяет схему образцов крови для получения данных о первой половине фармакокинетической кривой «концентрация-время». Схема отбора образцов должна быть составлена так, чтобы Сmax не являлась первой точкой на фармакокинетической кривой. Также необходимо обеспечить достаточную частоту заборов крови около предполагаемого tmax, как правило это должно быть 3 точки до ожидаемого tmax (исключая 0 точку) и 3 точки после. По крайней мере, 3 или 4 образца необходимы для достоверной оценки терминальной фазы элиминации. Т.е. минимальная схема отбора образцов крови может состоять из 10-11 точек, но как правило количество точек отбора крови превышает 18, что бы достаточно точно описать фармакокинетические профили сравниваемых препаратов.
Особенности лекарственной формы – данный фактор определяет не столько дизайн исследования, сколько в целом подход к изучению биоэквивалентности. Так для большинства парентеральных лекарственных не проводят фармакокинетические сравнительные исследования. Для препаратов диспергирующих в полости рта – это особенности приема, для препаратов подъязычных или защечных которые всасывают во рту это также особенности приема и график отбора проб крови. Для препаратов с модифицированным высвобождением это объем требуемых исследований для доказательства биоэквивалентности. Так для препаратов с обычным (немедленным) высвобождением как правило достаточно одного исследования для одной дозировки, то для препаратов с модифицированным высвобождением может потребовать 3 исследования (в условия приема препаратов натощак, с приемом пищи и с многократным приемом).
Кумуляция – если известно, что лекарственное вещество обладает кумуляцией, т.е. когда наблюдается существенное содержание в организме перед следующим приемом новой дозы, то требуется проведение исследования с многократным дозированием. То есть вместо одной дозы каждый доброволец (или пациент) принимает несколько доз исследуемого и референтного препарата и оцениваются фармакокинетические параметры в интервале дозирования. Например, в течение 3 дней каждый день доброволец или пациент принимает по 1 дозировке исследуемого препарата, затем после периода отмывки в течение 3 дней принимает по 1 дозировке референтного препарата.
Как правило исследования с многократным дозированием требуются для препаратов в лекарственных формах с модифицированным высвобождением, т.к. за счет модификации высвобождения часто увеличивается длительность экспозиции лекарства в организме и соответственно его накопление.
Наличие эндогенных концентраций – фактор, затрудняющий оценку содержания действующего вещества лекарственного препарата в крови после его приема, поэтому требуется вводить в исследование дополнительный этап для сбора крови перед приемом препаратов для определения эндогенного уровня.
Фармакокинетические половые различия – фактор, который вносит вклад в вариабельность фармакокинетики и может приводить к не биоэквивалентности препаратов, поэтому в некоторых случаях в исследования не включают женщин, если известны такие половые различия для конкретного лекарственного вещества. Вместе с тем такой подход вызывает сомнения, если препараты назначают для лечения и мужчин и женщин, т.к. подтверждённая биоэквивалентность только у мужчин не говорит о том, что в исследовании у женщин она была бы подтверждена. Поэтому все же лучше включать участников обоего пола в исследования биоэквивалентности и соблюдать их пропорциональное соотношение.
Фармакогенетические различия – данный фактор также может вносить вклад в оценку фармакокинетики если лекарственное вещество по-разному метаболизируется у субъектов с определенным генотипом. Поэтому если такие фармакогенетические различия известны и часто встречаются в популяции, то при скрининге рекомендуется проводить фармакогенетическое исследование, с целью невключения таких субъектов.
Итак, существуют две основные разновидности дизайна исследований биоэквивалентности, это перекрестный, когда каждый доброволец или пациент принимают и исследуемый и референтный препарат, или параллельный, когда каждый доброволец принимает либо исследуемый, либо референтный препарат.
У каждой из этих разновидностей есть свои плюсы и минусы.
Дополнительные разновидности перекрёстного дизайна
Для некоторых лекарств простой перекрестный дизайн может не подойти, т.к. в случае высокой внутрииндивидуальной вариабельности фармакокинетических параметров изучаемых в исследовании биоэквивалентности Cmax и AUC будет невозможно доказать биоэквивалентность на небольшом размере выборки, а то количество добровольцев или пациентов которое требуется для этого окажется столь большим, что целесообразность в проведении исследования биоэквивалентности по ФК конечным точкам будет равна 0. Потому что с таким размером выборки исследование выйдет очень дорогим и можно будет провести исследование по клиническим конечным точкам эффективности.
Принято, что высокая вариабельность считается, когда коэффициент внутрииндивидуальной вариабельности (CVintra) по результатам ранее выполненных исследований превышает значение 30%. В таблице ниже показан размер выборки в зависимости от коэффициента внутрииндивидуальной вариабельности (CVintra) и предполагаемой точечной оценки (PE) [данная оценка предполагает на сколько исследуемый препарат отличается от референтного].
В таких случаях должен использоваться повторный (репликативный) перекрестный дизайн, то есть, когда повторяется схема простого перекрестного дизайна. Выделяют полуповторный (частичный репликативный) и полный повторный (полный репликативный) варианты. При полуповторном схема повторяется не полностью, а только для референтного препарата, и исследование состоит из 3 периодов. При полном повторном схема повторяется полностью и поэтому исследование состоит из 4 периодов.
На схеме ниже приведены основные характеристики.
Еще одной разновидностью перекрестного дизайна является адаптивный (двухэтапный).
Данный дизайн предполагает также повторение схемы простого перекрестного дизайна на втором этапе исследования, в случае если биоэквивалентность не будет подтверждена после первого этапа по причинам не достаточной мощности исследования из-за недостаточного размера выборки.
Стоить отметить, что данный вид дизайна исследований часто применяется в России, а за рубежом более распространено выполнение пилотных исследований биоэквивалентности на небольшой выборке и затем при необходимости выполнение нового опорного исследования на большей выборке. Это является более приемлемым с регуляторной точки зрения, т.к. исключает риск ложноположительного заключения о биоэквивалентности и риск возможного манипулирования данными.
Исторически сложилось, что основными методами оценки результатов таких исследований являются методы B и C по D. Potvine. Именно данные методы сейчас часто применяются Российскими исследователями и принимаются регулятором.
Для того чтобы правильно выбрать дизайн для исследования биоэквивалентности следует в первую очередь оценить данные о лекарственном препарате (особенности действующего вещества его фармакокинетику, особенности лекарственной формы, наличие эндогенных концентраций); оценить данные о результатах ранее выполненных исследований биоэквивалентности (дизайн исследований, данные о внутрииндивидуальной/межиндивидуальной вариабельности, размере выборки), после чего определить тот дизайн, который будет подходить для конкретного лекарственного препарата исходя выше перечисленных факторов. Одновременно с планирование дизайна исследования также необходимо спланировать и статистический план исследования, т.к. они между собой взаимосвязаны.
Информацию и кейсы о статистическом планировании исследований биоэквивалентности ищи в ТГ канале bioequivalence_in_EAEU.