Сегодня мы обсудим патогенез и текущие терапии самых распространенных хронических болезней накопления (болезни Фабри, Гоше, Помпе и мукополисахаридозов), при которых подбор действительно эффективного лечения важен как никогда, однако возможности современной медицины (как мы увидим) далеко не всегда позволяют в полной мере это обеспечить.
Болезнь Фабри —
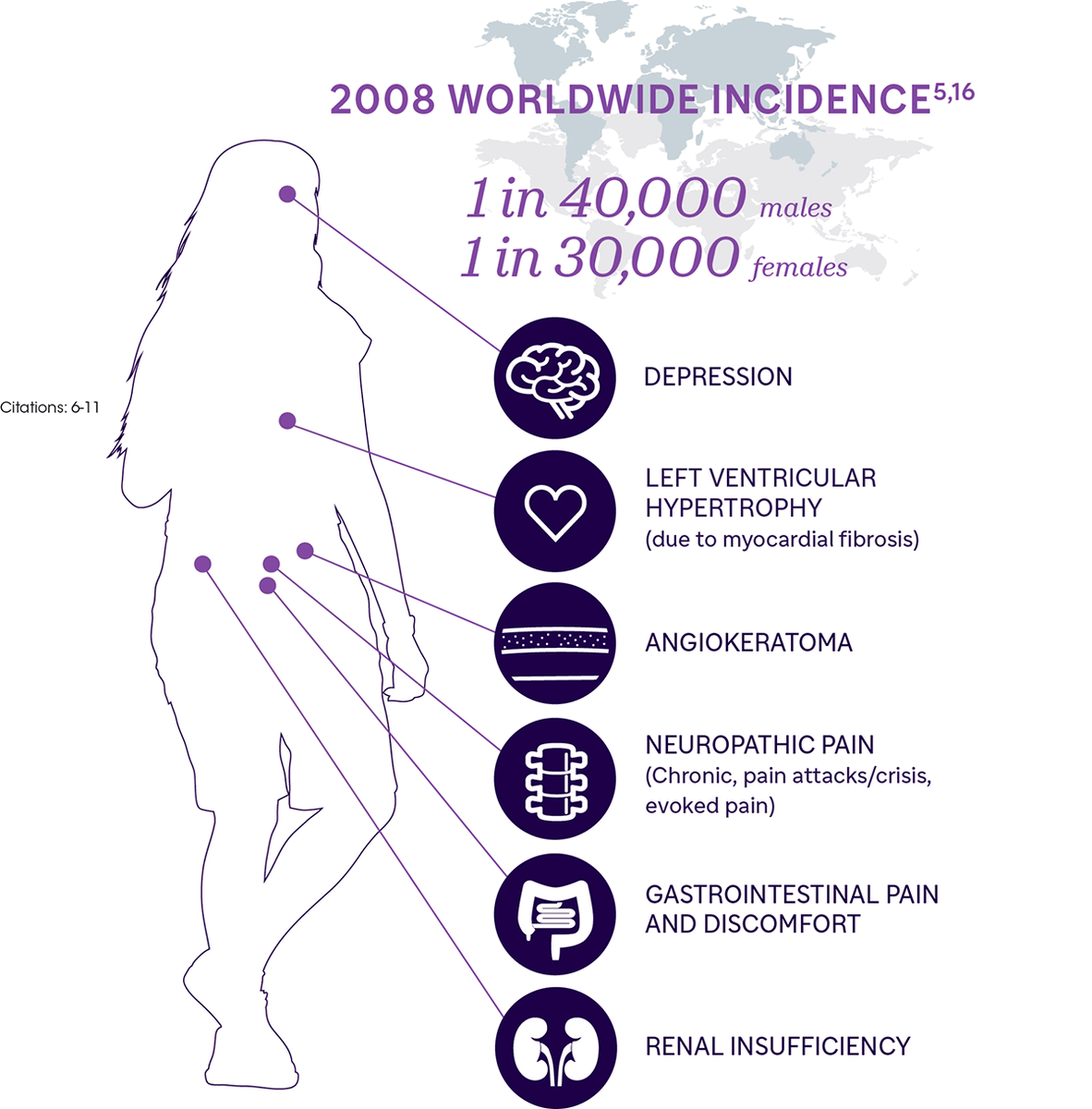
редкое заболевание накопления, при котором дефицит α-галактозидазы А (α-Gal A) приводит к накоплению глоботриаозилцерамида (GL-3) и некоторых других гликосфинголипидов в различных клетках. «Излишний» GL-3 может привести к прогрессирующему повреждению клеток, фиброзу, хронической боли, недостаточности органов и в конечном итоге (если всё это не лечить) к преждевременной смерти. Из органов в первую очередь затрагиваются почки, сердце, нервная система, но также описаны и респираторные заболевания, включая обструкцию дыхательных путей, интерстициальные заболевания легких и повышенную заболеваемость обструктивным апноэ во время сна (ОАС). Эти состояния прогрессируют, то есть со временем ухудшаются, хотя происходить это может медленно. У мужчин обычно возникают серьезные проблемы со здоровьем к 30–45 годам; у женщин проблем может не быть до 50 лет и намного старше.
В случае болезни Фабри ожидаемая продолжительность жизни во многом зависит от своевременности постановки диагноза и оперативности назначения соответствующего лечения (это верно и для других болезней накопления), хотя, увы, в среднем такие пациенты живут меньше, чем здоровые люди. Опубликованные для США данные показывают, что мужчины, страдающие болезнью Фабри, в среднем живут лишь 58 лет по сравнению с 75 годами для мужчин в общей популяции. Для женщин с болезнью Фабри средняя продолжительность жизни составляет уже 75 лет по сравнению с 80 годами для здоровых.
Кстати, на «Биомолекуле» в цикле «Орфанные заболевания» уже выходила статья про этот недуг: «Болезнь Фабри: частая среди редких» — читайте там больше об истории открытия, патогенезе и современном лечении, в том числе разрабатываемом в России.
Основой терапии болезни Фабри является ФЗТ. В Соединенных Штатах для этого довольно давно (еще в 2003 г.) одобрена агалсидаза бета (Fabrazyme) , а уже совсем недавно (в 2023 г.) там же получил одобрение препарат Elfabrio: это пегилированная форма агалсидазы альфа. Пегилирование продлевает «жизнь» фермента: пациенты могут реже делать утомительные инъекции, что повышает качество жизни и приверженность лечению. Обычная же (не пегилированная) форма агалсидазы альфа — это препарат Replagal, коммерчески доступный во многих странах (но не в США), включая Канаду, Россию, Великобританию, Мексику, Израиль и весь Евросоюз.
Кстати биосимиляр данного лекарства (под коммерческим названием «Фабагал») планирует производить российская компания «Петровакс»: в 2023 году они получили на него регистрационное удостоверение, а уже на конец 2024 года планируют запуск производства полного цикла (включая субстанцию) для выпуска коммерческих серий.
К сожалению, ФЗТ при болезни Фабри трудно назвать идеальной — она купирует далеко не все симптомы: патологические процессы нарушения функции почек и гипертрофии левого желудочка могут продолжаться; кроме того, существенным недостатком являются возможные аллергические реакции на инъекции, в том числе тяжелые. Так или иначе, правильный протокол лечения этой болезни зачастую требует применения нескольких препаратов в составе комплексной терапии.
Одна из возможных альтернатив ФЗТ — мигаластат (Galafold компании Amicus Therapeutics): фармакологический шаперон, то есть низкомолекулярное соединение, которое связывается с активным сайтом мутантного фермента альфа-галактозидазы и стабилизирует его. Это лекарство принимается перорально и потому подходит пациентам, не переносящим ФЗТ из-за образования нейтрализующих антител к вводимому ферменту, аллергических реакций, связанных с инфузией, или по каким-либо другим причинам. К сожалению, эта альтернатива помогает далеко не всем: мигаластат работает, если у больного мутации приводят к неправильному сворачиванию альфа-галактозидазы; в том же случае, если этот фермент совсем отсутствует, фармакологический шаперон будет бессилен. Считается, что только примерно у 35–50% пациентов с болезнью Фабри могут быть варианты мутаций, поддающиеся лечению мигаластатом.
Болезнь Гоше —
возникает в результате недостатка лизосомального фермента глюкоцереброзидазы и вытекающего накопления его субстрата глюкоцереброзида. С течением болезни этот и другие гликолипиды начинают накапливаться в лизосомах макрофагов, что приводит к разным нарушениям.
В какой-то момент «нагруженные» липидами макрофаги скапливаются в селезенке, печени, костном мозге, костях, других тканях и органах и вызывают их системное поражение: гематологические нарушения, увеличение органов, повреждения скелета. В зависимости от наличия или отсутствия неврологических поражений описаны три клинических подтипа болезни Гоше:
- I тип — без неврологических нарушений, это наиболее распространенная форма заболевания. Его клиническая картина варьирует от бессимптомного течения на протяжении всей жизни до форм патологий, проявляющихся еще в детстве. В последнем случае замедляются рост и половое созревание, увеличиваются печень и селезенка, возникают аномалии костей; болезнь Гоше I типа может вызвать серьезные заболевания печени, включая повышенный риск кровотечения в желудке и пищеводе и рак печени. Симптомы этой болезни могут проявиться в любом возрасте, но средний возраст больных в момент манифестации — 30 лет. Современное лечение этого недуга (см. ниже) довольно эффективно и при его корректном применении позволяет рассчитывать на почти нормальную продолжительность жизни.
- II тип встречается реже и характеризуется ранним и тяжелым поражением центральной нервной системы, также увеличением печени и селезенки, что приводит к смерти в возрасте до трех лет; в настоящее время от этого заболевания нет лекарства. Текущая терапия в основном паллиативная: поддерживающий уход с вниманием к вопросам, общим и для других нейродегенеративных состояний (таким как питание); а также и к более специфичным для данного недуга проблемам: риску аспирации дыхательных путей, необходимости респираторной поддержки, и пр.
- III тип также называют ювенильной, или подострой неврологической болезнью Гоше: для нее характерны внутренние симптомы, описанные для болезни Гоше I типа, — они часто развиваются на первом году жизни, обычно в сочетании с неврологическими глазодвигательными нарушениями, проявляющимися позднее — в возрасте 6–15 лет. Продолжительность жизни больных болезнью Гоше III типа, увы, невелика: 12–17 лет, в редких случаях — 30–40 лет.
Лечение болезни Гоше I и III типов состоит либо из ФЗТ, либо из субстрат-снижающей терапии (ССТ). Существует три одобренных FDA препарата ФЗТ: имиглюцераза (Cerezyme), велаглюцераза альфа (VPRIV) и талиглюцераза альфа (Elelyso); и два одобренных FDA препарата ССТ: миглустат (Zavesca) и элиглустат (Cerdelga).
ССТ работает путем ингибирования фермента глюкозилцерамидсинтазы, блокируя первый этап в синтезе гликосфинголипидов, тем самым ограничивая выработку глюкоцереброзида. Одно из преимуществ ССТ — пероральное, а не внутривенное введение. Клиническая практика сильно различается, но большинство вновь диагностированных пациентов начинают с ФЗТ. Некоторые переходят на ССТ в более позднем возрасте. Детям с болезнью Гоше следует начинать ФЗТ, как только у них появляются симптомы. Талиглюцераза альфа, миглустат и элиглустат одобрены только для использования у взрослых. Как ФЗТ, так и ССТ по итогу применения увеличивают количество тромбоцитов и концентрацию гемоглобина, уменьшают объем печени и селезенки и снижают биомаркеры, характерные для болезни Гоше (такие как хитотриозидаза) [13].
Несмотря на то, что ФЗТ и ССТ улучшают гематологические показатели и состояние внутренних органов, они, по-видимому, не влияют (или мало влияют) на неврологические нарушения. Теоретически болезни I и III типа можно также вылечить с помощью трансплантации гемопоэтических стволовых клеток, но это считается крайней мерой, поскольку может привести к смерти или инвалидности.
Болезнь Помпе —
прогрессирующее, мультисистемное, изнурительное и часто опасное для жизни нервно-мышечное расстройство. Вызывается она отсутствием или дефицитом фермента кислой альфа-глюкозидазы (GAA), отвечающим за расщепление гликогена внутри клеток. Без достаточного количества этого фермента гликоген накапливается в первую очередь в мышечных клетках (также в гепатоцитах, эндотелиальных клетках, нейронах ЦНС), что в какой-то момент неминуемо нарушает мышечную функцию. Выходит так потому, что прогрессирующее накопление гликогена приводит к разрыву лизосомальных мембран, вызывая утечку гидролитического материала в цитоплазму различных мышечных (и что особо значимо — скелетных и сердечных) клеток с нарушением их сократимости. Всё это приводит к мышечной слабости, истощению: во многом за счет поражения расположенных ближе к центру тела (проксимальных) мышц и легочной функции болеющих людей.
Младенцы, у которых почти полностью отсутствует активность кислой альфа-глюкозидазы (менее 1% от нормы), — это пациенты с классической (инфантильной или младенческой) болезнью Помпе (МБП), и участь их наиболее печальна. Симптомы этого недуга манифестируют в среднем между 1,6 и 2 месяцами жизни (хотя могут проявляться даже до рождения, когда ребенок еще находится в матке), и до появления ФЗТ такие дети чаще всего умирали в первый год жизни от сердечной недостаточности.
Но и сейчас выжившим благодаря лечению часто требуется респираторная поддержка, включая искусственную вентиляцию легких, зонды для кормления, ходунки и/или инвалидные коляски. При этом смертность таких маленьких пациентов всё равно очень высока (в разном возрасте различна), и невелика их общая продолжительность жизни. Для получающих лечение самыми сложными оказываются первые три года жизни, так как в этот период умирают очень многие (по некоторым данным около 50%); а следующий пик снижения выживаемости приходится на возраст уже после 12 лет. Оценить средний возраст выживаемости при МБП пока довольно сложно, так как статистики не хватает.
У лиц с болезнью Помпе с поздним началом (БППН) активность фермента может составлять 40% или менее от нормы, а симптомы проявляются в любом возрасте: от детства до взрослой жизни. Как правило, чем раньше началась болезнь, тем тяжелее она протекает и быстрее прогрессирует. Если БППН проявилась уже во взрослом возрасте, нормальная функция сердца таких пациентов может сохраняться, и они могут страдать лишь от слабости проксимальных мышц.
Соответственно, продолжительность жизни людей с БППН также сильно варьирует: смерть может наступить в период от раннего детства до старости, на что влияют характер течения недуга, наличие респираторных проблем, других сопутствующих заболеваний и т.д. Происходит так во многом потому, что заболевание патофизиологически очень гетерогенно, что вызывает разнообразие клинических проявлений. Из-за отсутствия данных (ФЗТ стала доступна только около 15 лет назад) всё еще довольно сложно точно оценить влияние современных методов лечения на продолжительность жизни пациентов с БППН.
Текущее лечение: неполный триумф ФЗТ
Препараты ФЗТ, содержащие GAA (препараты Myozyme, Lumizyme) когда-то радикально изменили специфику прогноза при МБП. Связано это с тем, что ФЗТ очень хорошо помогает при «сердечных» осложнениях этой болезни: снижает гипертрофическую кардиомиопатию с улучшением и/или нормализацией индекса массы левого желудочка и других «сердечных параметров» (иногда в течение всего нескольких недель после начала такой терапии); после этого аритмии возникают уже у меньшего числа пациентов. Благодаря всему этому сердечные осложнения при МБП уменьшились, выживаемость повысилась, однако это хроническое расстройство всё еще идет с тяжелыми нервно-мышечными отклонениями (скелетные мышцы плохо реагируют на ФЗТ) и до сих пор летально, хотя пациенты и живут зачастую немного дольше.
91% пациентов с МБП, получавших лечение с помощью ФЗТ, имеют слабость и гипотонию, 91% — дизартрию, 45% — дисфагию, 36% — зависимость от зонда для кормления, 36% нуждаются во вспомогательных устройствах для передвижения, и многим из них необходим дополнительный кислород: искусственная вентиляция легких при дыхательной недостаточности и ОАС. У пациентов с БППН всё уже «получше»: у них стабилизируется функция дыхания, улучшается моторика, поддерживается способность передвигаться и увеличивается выживаемость. На фоне проводимой ФЗТ такие пациенты могут дожить до старости с хорошими показателями качества жизни.
К сожалению, при всех плюсах ФЗТ нужно признать, что для пациентов с болезнью Помпе это далеко не панацея: из-за крайне сложного патогенеза им зачастую требуется многопрофильная помощь.
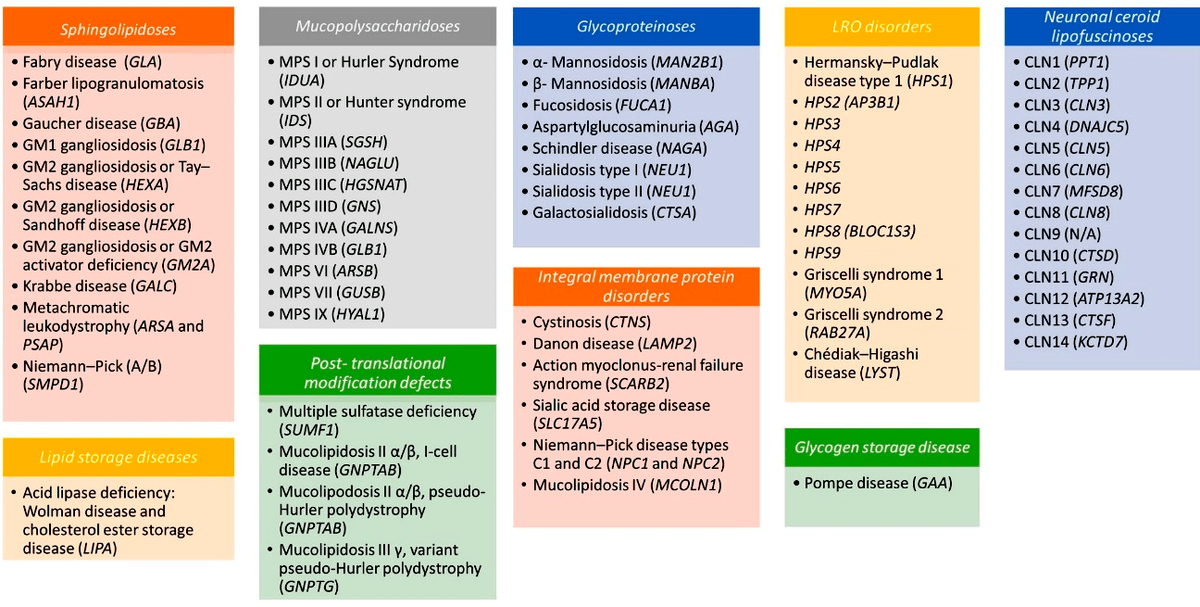
Мукополисахаридозы (МПС) —
это ЛБН, вызванные дефицитом лизосомных гидролаз, участвующих в деградации гликозаминогликанов (ГАГ). В настоящее время известно десять различных типов МПС, и каждый из них может «являться миру» в широком диапазоне клинической тяжести. Например, при МПС I самая тяжелая форма называется синдромом Гурлер; промежуточная по тяжести — синдромом Гурлер—Шейе; а наименее тяжелая — синдромом Шейе. При этом МПС I и II — это наиболее распространенные типы мукополисахаридозов. Клинические проявления всех этих заболеваний различаются в зависимости от типа «дефицитного» фермента и степени его остаточной активности; однако симптомы их обычно прогрессируют — это грубые черты лица, гепатоспленомегалия, пороки клапанов сердца, респираторные заболевания и аномалии костей/суставов.
Первичное поражение ЦНС присутствует при большинстве МПС, особенно при тяжелых формах I–III и VII типов, причем проявляется всё это в том числе снижением интеллекта, зачастую грубым, прогрессирующим по мере развития заболевания. Дисфункции ЦНС, как правило, начинают проявляться на 2–3 году жизни, что в конечном итоге приводит к потере освоенных маленькими пациентами нейромоторных навыков. Эти разрушительные симптомы неумолимы в своем ухудшении, приводя зачастую к преждевременной смерти.
Собственно смертность, связанная с этими болезнями, часто обусловлена как раз тяжелой нейродегенерацией, респираторными инфекциями и дисфункциями сердца. Прогноз при этом почти всегда неблагоприятный, ведь мукополисахаридозы характеризуются катастрофическими системными нарушениями. Продолжительность жизни тут невелика (хоть для разных форм и различна): от 6–10 лет при синдроме Гурлер до 30–40 лет при менее тяжелых формах.
Лечение этих болезней в настоящее время основано главным образом на применении ФЗТ, а также трансплантации гемопоэтических стволовых клеток. Используемые ФЗТ ныне это: ларонидаза (Aldurazyme) для МПС I, идурсульфаза (Elaprase) для МПС II и вестронидаза альфа (Mepsevii) при лечении МПС VII. Такие лекарства способны замедлять прогрессирование этих заболеваний, но не излечивают, а поскольку вводимые ферменты не проникают через ГЭБ, они также мало что дают в плане облегчения неврологических симптомов (которые, очевидно, здесь являются одной из основных проблем).
Трансплантация гемопоэтических стволовых клеток может быть проведена у пациентов с МПС I в возрасте до двух лет , и хороша она как раз тем, что способна стабилизировать нейрокогнитивную функцию за счет восстановления микроглии и соответствующей нормализации активности лизосомных ферментов в мозге. Вот почему эта процедура — часто единственный способ значительно снизить клинические проявления тяжелых форм МПС I (особенно в части поражения ЦНС). К сожалению, при всех её плюсах, эффективность всё же ограничена, неся при этом крайне высокие риски тяжелых осложнений и летальности, и показана пациентам далеко не всегда.
Ранняя трансплантация лучше тем, что симптомы заболевания еще не столь остры, а потому можно надеяться на неплохую эффективность.
В меньшей степени доказана польза этого метода при МПС II.
Больше о лизосомных болезнях накопления читайте на нашем сайте!