Секвени́рование (белков и нуклеиновых кислот — ДНК и РНК) — определение их аминокислотной или нуклеотидной последовательности (от лат. sequentum — последовательность).
Если по человечески, то секвенирование – это изучение генома с целью выявления “семейных” заболеваний и предрасположенностей. С этими знаниями можно подобрать действительно индивидуальную терапию.
Краткий обзор
Для того, чтобы правильно интерпретировать результаты диагностического теста секвенирования набора генов, необходимо учитывать генное покрытие, взятый для рассмотрения. Если покрытие генов слишком низкое, то необходимо повторное секвенирование, чтобы убедиться, что патогенные изменения не пропущены. Чтобы облегчить интерпретацию данных генного покрытия, мы разработали CovReport – новый и простой инструмент визуализации. Он генерирует краткую сводку генного покрытия, которая позволяет с первого взгляда оценить результативность теста секвенирования. Покрытие как на уровне генов, так и на уровне экзонов может быть немедленно оценен и принят во внимание для принятия дальнейших медицинских решений. CovReport не требует сложной установки и может быть легко реализован в любой диагностической лаборатории. Удобный интерфейс генерирует графическую сводку покрытия, которая может быть включена в диагностический отчет. В дополнение к автономной версии мы также предоставляем версию CovReport с командной строкой, которая может быть интегрирована в любой биоинформационный конвейер. Этот гибкий инструмент теперь является частью обычного анализа секвенирования в отделении медицинской генетики больницы Ла-Тимон (Марсель, Франция). CovReport доступен по адресу http://jdotsoft.com/CovReport.php. Он реализован на Java и поддерживается в Windows, Mac OS X и Linux.
Предисловие
С момента появления технологий секвенирования по методу картирования коротких прочтений были затрачены значительные усилия на повышение качества полученных данных с целью удовлетворения требований молекулярной диагностики. Однако многие области человеческого генома по-прежнему трудно анализировать с помощью данного метода секвенирования. Эти "темные" участки генома содержат ряд генов, которые отвечают за болезни человека1. Например, несколько нервно-мышечных болезнетворных генов, таких как NEBULIN (NEB) и SELENON (SEPN1), перекрывают эти трудные для секвенирования области. Болезнетворные изменения в области генов с субоптимальным покрытием последовательностей могут быть упущены из виду. Таким образом, при проведении коротких прочтений секвенирования в диагностической установке, выявление областей с плохим покрытием имеет решающее значение для интерпретации результата диагностического секвенирования. Если покрытие недостаточно даже для небольшого участка подозреваемого гена, необходимо повторно провести тест секвенирования, чтобы убедиться, что патогенный вариант не пропущен. Чтобы облегчить интерпретацию данных покрытия молекулярными генетиками и теми, кто назначают генетическое тестирование, мы разработали новый и простой в использовании инструмент визуализации - CovReport. Краткая сводка данных о покрытие, генерируемая данным инструментом, позволяет с первого взгляда оценить эффективность теста секвенирования.
Методология
Развертывание / Внедрение / Установка
CovReport реализован как автономное приложение Java. Он может работать на любой платформе, где установлена Java Runtime (JRE) (Windows, Mac OS и Linux). Поддерживаемая версия Java - 8. Приложение использует внешние библиотеки с открытым исходным кодом (JARs), которые встроены в основной исполняемый файл JAR. Используются следующие внешние библиотеки:
· Apache PdfBox (https://pdfbox.apache.org),
· Apache CLI (http://commons.apache.org/proper/commons-cli),
· JarClassLoader (http://www.jdotsoft.com/JarClassLoader.php).
Приложение можно загрузить с сайта http://www.jdotsoft.com/CovReport.php в виде архива CovReport.zip. После извлечения на локальный диск папка содержит следующее:
· msg – папка с файлами сообщений интернализации, используемыми для генерируемых PDF-файлов,
· CovReport.jar – исполняемый архив Java,
· run.cmd – запуск приложения в Windows,
· runFromCommandLine.cmd – запуск с использованием командной строки Windows.
После первого запуска CovReport создаются следующие элементы:
· pdf-results – папка с PDF-файлами, создаваемыми приложением,
· CovReport.config – файл конфигурации; этот файл может быть вручную изменен и использован для замены значений по умолчанию для выполнения с помощью командной строки.
Приложение может быть запущено с пользовательским интерфейсом или из командной строки. Параметры командной строки:
· -i, – input < arg> входной файл
· -n, – name <arg> имя пациента
· -c, – config <arg> файл конфигурации (опционально)
Входной файл, содержащий информацию о покрытии каждого экзона, представляет собой CSV-файл, где данные разделены символом табуляции. Все записи в файле могут указываться как в кавычках, так и без них. Во входном файле не должен содержать никаких специальных символов. Ожидается наличие следующие столбцов в файле: RefSeqName, GeneSymbol, Exon, Size, Mean Depth, SD Depth, Coverage 1×, Coverage 5×, Coverage 10×, Coverage 20×, Coverage 30×. Пример заполненного входного файла приведен в разделе “Дополнительные данные” (также доступен по адресу http://www.jdotsoft.com/CovReport/44genes-stat_coverage_exon.txt). Входной файл может быть легко сгенерирован модулем покрытия VarAFT tool 2 (https://varaft.eu/), который использует BEDTools 3,4 для расчета покрытия на уровне экзона. Так результаты любого другого конвейера расчета покрытия могут быть преобразованы в соответствующий формат и использованы в качестве входных данных в CovReport. Версия CovReport для командной строки позволяет интегрировать этот инструмент визуализации в автоматизированный конвейер в качестве дополнительного шага анализа диагностических последовательностей.
Пользовательский интерфейс представлен на английском языке, но генерируемые PDF-файл будут сохраняться с учетом локальных языковых настроек. В текущем дистрибутиве поддерживаются английский и французский языки, но есть возможность добавления пользователем других языков. Текст, в генерируемых pdf-отчетах, также может быть настроен, что позволяет встроить CovReport в любую диагностическую установку.
Подробные инструкции по загрузке и запуску о CovReport описаны по адресу http://www.jdotsoft.com/CovReport.php.
Результат / Итог / Выводы
CovReport позволяет с первого взгляда оценить покрытие экзона для диагностического секвенирования, генерируя небольшой простой отчет в формате PDF. Интуитивно понятный интерфейс и отсутствие сложных этапов установки делает CovReport простым в применении для пользователей без специальной подготовки по биоинформатике. Возможность запуска CovReport с помощью командной строки позволяет встраивать его в любой биоинформационный конвейер.
Некоторые особенно полезные функции CovReport:
Во-первых, приложение работает на локальном компьютере, что обеспечивает безопасность данных,
Во-вторых, имя пациента из предыдущего анализа автоматически сбрасывается при вводе нового файла,
В-третьих, формат, содержание и язык отчета могут быть легко настроены лабораторией, адаптируясь к любой диагностической установке,
В-четвертых, информация о дополнительном повторном секвенировании Сэнгера не оптимально покрытых областей может быть интегрирована в отчет о покрытии первоначального секвенирования по методу коротких прочтений, что позволяет легко отслеживать эксперименты по секвенированию.
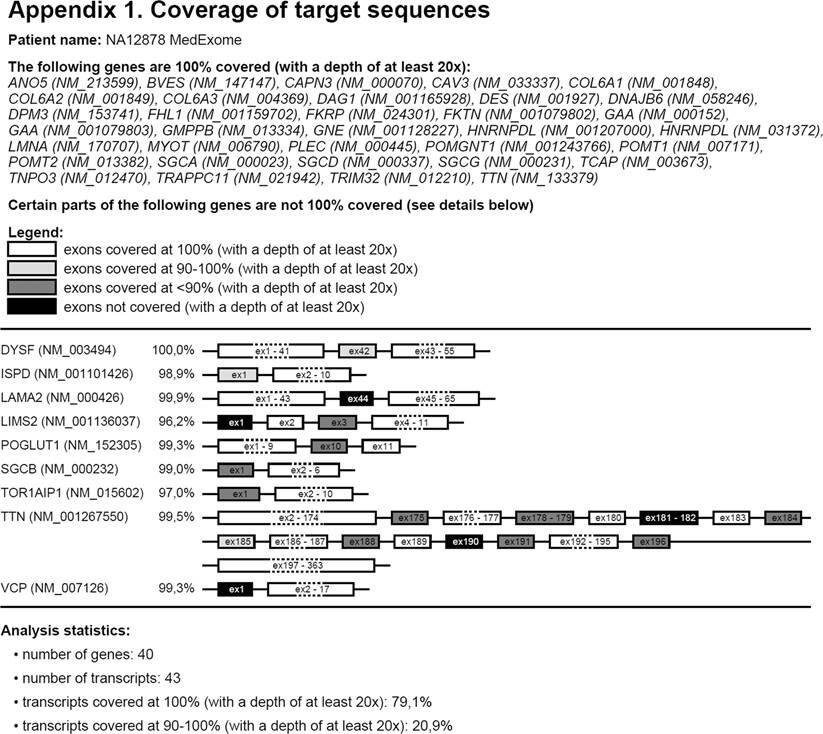
Покрытие экзонов для генов на экране визуализируется путем рисования экзонов, заштрихованных в соответствии с уровнем покрытия: 100% покрытые экзоны белые, 90-100% экзоны светло-серые, <90% покрытые экзоны темно-серые, не покрытые экзоны черные. Несколько вариантов визуализации позволяют адаптировать графическое представление к потребностям пользователя. Гены со 100% охватом могут быть перечислены в верхней части отчета без рисования экзонов (опция пропустить белые гены, по умолчанию). Схематичные структуры генов / экзонов будут нарисованы для остальных генов, затеняя экзоны с более низким покрытием. Если выбран вариант слияния белых экзонов, CovReport будет объединять экзоны, покрытые на 100% для более компактного представления, что полезно для генов с многочисленными экзонами. Если снять этот флажок, то будет создан отчет с каждым отдельным извлеченным экзоном. Аналогично, экзоны с одинаковым затенением могут быть объединены для более компактного представления (опция слияния небелых экзонов). Среднее покрытие генов может быть показано рядом с каждым геном (опция Show gene weighted coverage), который также используется для статистики общего покрытия панели генов (опция Show statistics). Информация о транскрипте RefSeq может быть включена в отчет (опция Show gene transcripts), рекомендованный, если транскрипты различаются по числу экзонов, что приводит к различиям в охвате между изоформами. Глубина покрытия по умолчанию 20x в отчете может быть изменена на 1×, 5×, 10× или 30×. Наконец, в отчет могут быть включены дополнительные замечания.
Статья переведена каналом "Голова руки". Оригинал статьи вы можете прочитать по ссылке.
Если понравилась статья, то Вы можете поставить лайк и подписаться на канала! (=