Речь пойдет об очень редком заболевании — метахроматической лейкодистрофии (МЛД). Однако революционный подход, который уже показал эффективность в ее лечении, может быть распространен и на более частые болезни. Теперь важно, чтобы последующие клинические данные не обманули ожиданий.
Что такое метахроматическая лейкодистрофия?
Метахроматическую лейкодистрофию еще называют сульфатидным липидозом. Это одна из многочисленных так называемых лизосомных болезней накопления. Их общая черта в том, что в результате генетического дефекта у человека нарушается нормальная работа одного из ферментов, отвечающих за превращения веществ в лизосомах.
В зависимости от того, какой фермент «сломан», в лизосомах накапливаются разные соединения, и при этом поражаются разные органы.
При этом заболевании не работает (или недостаточно активен) фермент арилсульфатаза А, которая катализирует отщепление сульфата от цереброзидсульфата с образованием цереброзида — главного гликолипида миелина.
Чаще всего заболевание развивается как следствие мутаций в гене ARSA. В настоящее время известно более 60 мутаций ARSA, которые приводят к нарушениям различной тяжести в зависимости от снижения активности фермента.
Что же это за нарушения и как они проявляются?
Фермент ARSA в норме больше всего активен в клетках, которые вырабатывают миелин. При дисфункции ARSA в лизосомах этих клеток накапливается цереброзидсульфат (или сульфатид, откуда второе название болезни — сульфатидный липидоз), что в итоге приводит к их гибели.
Поэтому все это в первую очередь приводит к разрушению белого вещества (миелина). Основные проявления — неврологические: в первую очередь задержка умственного развития и нарушения моторных навыков.
Также часто развиваются потеря чувствительности конечностей (периферическая нейропатия), недержание мочи, судороги, паралич, потеря речи, слуха и зрения.
Начало болезни, ее течение и прогноз зависят от возраста наступления болезни. У 50–60% пациентов симптомы наступают на втором году жизни. Для них характерна прогрессирующая слабость мышц, потеря способности ходить и стоять; затем, наоборот, мышцы становятся ригидными. Такие пациенты не доживают до взрослого возраста.
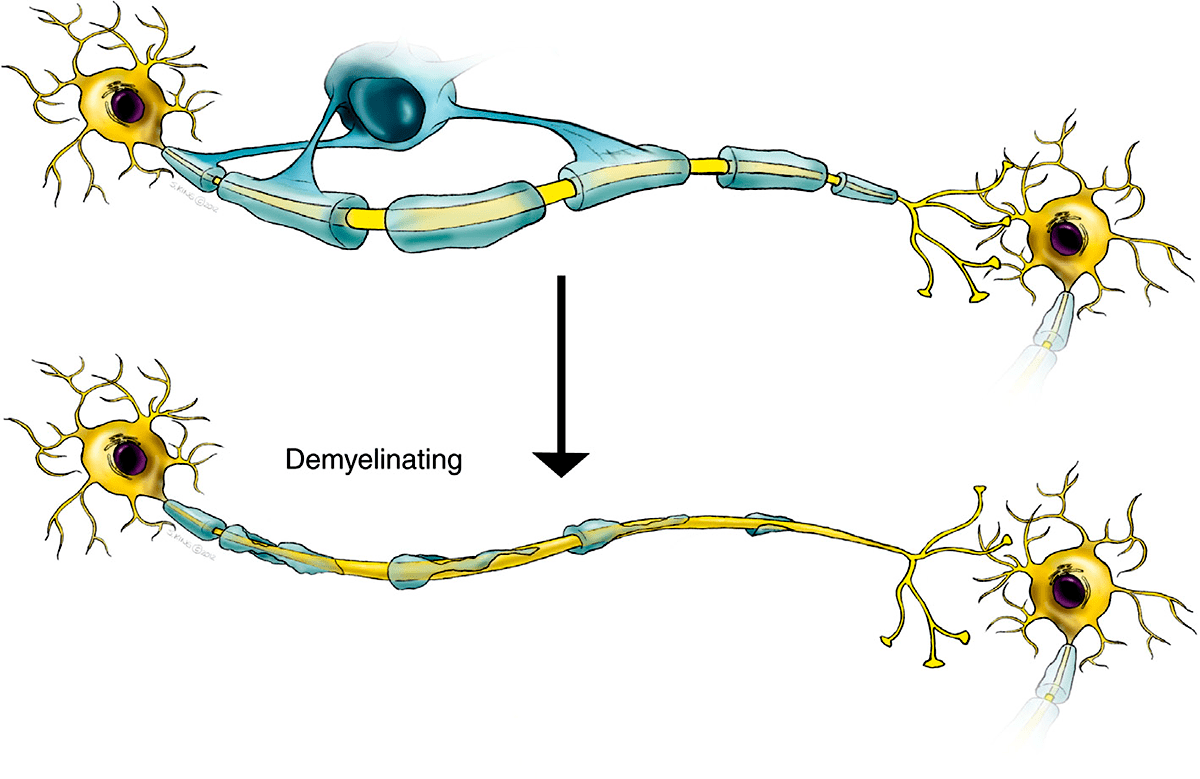
У 20–30% пациентов болезнь наступает после четырех лет. Симптомы развиваются медленнее, чем при младенческой форме, и пациенты, как правило, живут около 20 лет после постановки диагноза. Болезнь начинается с проблем в школе — ухудшается поведение и успеваемость. У 15–20% пациентов болезнь наступает в юности, первые симптомы — алкоголизм, наркомания, часто присоединяются галлюцинации.
Частота заболеваемости колеблется от одного человека на 40 000 до одного на миллион в зависимости от популяции, в среднем примерно один на 100 000. Таким образом, в России должно быть примерно 1400 таких больных, а в США — около 3200.
Лечение МЛД
В отличие от других болезней лизосомного накопления, для МЛД нет ферментозаместительной терапии (и вообще никакой одобренной специфической терапии).
Большинство пациентов получает только симптоматическую терапию (например, противоэпилептические препараты в случае судорог, мышечные релаксанты или физиотерапию), которая, хотя и облегчает их состояние, но не влияет на причину и патогенез болезни, и поэтому не сказывается на скорости ее прогрессии.
И тут на сцену выходит передовая терапия: подход, объединяющий генную и клеточную терапии.
Лентивирусы и клетки пациента
Попытки лечения МЛДметодами генной терапии предпринимаются с 1990-х годов. Единственное зарегистрированное исследование, проведенное всего на пяти пациентах, не имеет опубликованных результатов.
Итальянские ученые сделали выбор в пользу генной терапии ex vivo с использованием аутологичных клеток (полученных от самого пациента).
Исследователи отбирали гемопоэтические стволовые клетки костного мозга пациентов и трансдуцировали их лентивирусным вектором, несущим нормальный ген ARSA.
Лентивирусный вектор (безопасное производное вируса иммунодефицита человека) обеспечивает встраивание гена, который он несет, в геном клетки, которую заражает. Перед переносом генномодифицированных клеток пациенту проводят химиотерапию для уничтожения его собственных кроветворных стволовых клеток.
Как было показано и у мышей, и у людей, генномодифицированные стволовые клетки проникают за гематоэнцефалический барьер и секретируют там ARSA. За счет наличия остатков манноза-6-фосфата на поверхности ARSA проникает в лизосомы клеток нервной системы и обеспечивает нормальное превращение цереброзидсульфата в цереброзид.
В 2013 году опубликовали первые клинические данные на трех пациентах в возрасте 7–15 месяцев. В течение 18–24 месяцев после терапии у них сохранялись генномодифицированные клетки и наблюдалось достаточное число копий вектора на клетку и высокий уровень ARSA. Но что самое главное, у пациентов полностью затормозилось развитие симптомов и прогрессия поражений мозга на МРТ.
В сентябре 2019 года были опубликованы свежие данные. Сейчас период наблюдения за первым пролеченным пациентом уже превысил восемь лет, и он развивается так же, как и его нормальные сверстники. Всего пролечено 29 пациентов, из них терапия помогла 25-и. На рис. 5 приведены данные по умственному развитию пациентов, пролеченных в младенческом возрасте.
Процедура оказалась абсолютно безопасной, и, главное, ни у одного из пациентов не было отмечено признаков репликации вируса, клональной экспансии (превалирование какого-либо из клонов стволовых клеток, которое может свидетельствовать о том, что встраивание лентивируса дало клетке пролиферативное преимущество) или злокачественных заболеваний.
В первой половине 2020 года компания Orchard Therapeutics, которая коммерциализирует эту разработку, планирует подать пакет документов для разрешения на продажу в Европейское медицинское агентство.
Заключение
На наших глазах разворачивается потрясающая картина, когда болезни, ранее считавшиеся неизлечимыми и смертельными, удается вылечить, возможно, насовсем. Да, пока что это заболевания, затрагивающие максимум тысячи пациентов, а само лечение стоит почти всегда ошеломляюще дорого.
Однако лиха беда начало: по мере того, как ученые всё лучше овладевают навыками создания безопасных и эффективных методов терапии на основе генномодифицированных клеток, нам станут наверняка подвластны и более распространенные заболевания, а цена таких «клеточных лекарств» неизбежно снизится.
Больше подробностей о МЛД и лечении этого заболевания можно найти тут. Биомолекула благодарит вас за то, что вы прочитали эту статью до конца.
Будем рады вашим лайкам и подписке на наш канал —здесь мы рассказываем много интересного из мира науки! 💚
