1. Введение: Почки как высокоэнергетическая система и последствия метаболического коллапса
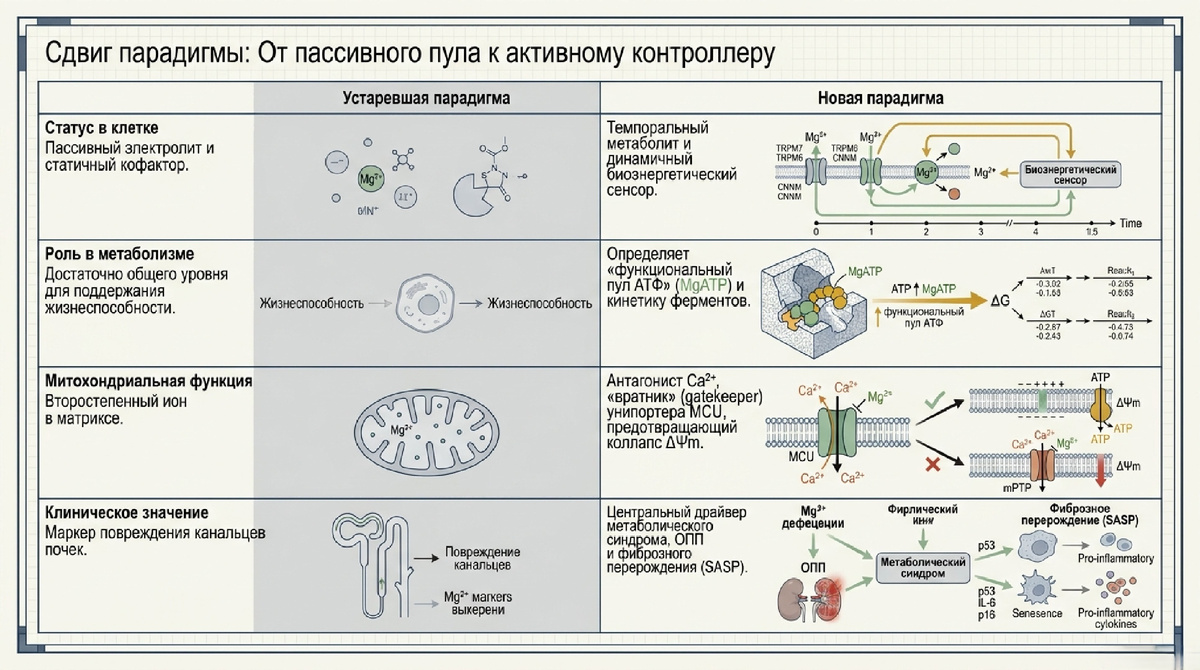
Почки представляют собой одну из наиболее метаболически напряженных систем организма, функционирующую на «преципитате гипоксии». Обладая массой менее 1% от общей массы тела, этот орган потребляет 20–25% покоящегося кислорода, что сопоставимо по интенсивности метаболизма только с миокардом. Данный термодинамический подвиг продиктован физиологическим мандатом на непрерывный транскластерный транспорт электролитов. Эпителиальные клетки проксимальных канальцев (PTEC) являются облигатными аэробами, чья жизнеспособность напрямую зависит от плотности митохондриального пула, обеспечивающего энергией насосы Na+/K+-АТФазы. Следовательно, любая дестабилизация ионного микроокружения неизбежно ведет к декомпенсации всей системы.
В патогенезе перехода острого повреждения почек (ОПП) в хроническую болезнь почек (ХБП) критическим событием является дезадаптивное метаболическое перепрограммирование. Изначально адаптивный «эффект Варбурга» — переключение на аэробный гликолиз при ишемии — в долгосрочной перспективе приводит к персистирующему подавлению окисления жирных кислот (FAO). Это провоцирует липотоксичность, разобщение окислительного фосфорилирования (OXPHOS) и формирование профибротического секреторного фенотипа, ассоциированного со старением (SASP). Традиционные нутритивные модели, сфокусированные исключительно на субстратах («топливе»), являются неполными без учета «машинерии» — ионного гомеостаза, где магний выступает фундаментальным регулятором митохондриальной эффективности.
2. Молекулярная архитектура транспорта магния: От нефрона к митохондрии
Транспорт Mg2+ в почках организован как иерархическая релейная система, преобразующая системные градиенты во внутриклеточные сигнальные каскады.
- Апикальный вход (Heterotetrameric Gateway): Основным порталом реабсорбции в дистальном извитом канальце (DCT) является комплекс «чанзимов» TRPM6/TRPM7. Согласно данным криоэлектронной микроскопии, функциональный канал в DCT представляет собой гетеротетрамер, где уникальная архитектура TRPM6 обеспечивает тонкую настройку входа катионов. Мутации TRPM6 манифестируют в виде тяжелой системной гипомагниемии, которую невозможно компенсировать гомомерами TRPM7, что подтверждает иерархическое превосходство TRPM6 в почечном гомеостазе.
- Базолатеральная экструзия и модуль CNNM2–PRL: Выход магния в интерстиций осуществляется через белок CNNM2, содержащий домены DUF21 и CBS. Этот процесс регулируется фосфатазами регенерирующей печени (PRL). Связывание PRL с доменом CBS CNNM2 изменяет конформационный ландшафт транспортера, подавляя экструзию и способствуя внутриклеточной кумуляции Mg2+ для поддержания метаболической компетенции.
- Митохондриальный интерфейс и электрофоретический транспорт: Матриксный пул Mg2+ управляется динамическим балансом между MRS2 и SLC41A3. Канал MRS2 обеспечивает электрофоретический вход Mg2+, движимый мембранным потенциалом (ΔΨm). Важно отметить, что MRS2 не является абсолютно селективным: он способен проводить Ca2+ и другие катионы, а его активность лимитируется состоянием канала, а не только концентрационным градиентом. Напротив, белок SLC41A3 выступает в роли системы митохондриального эффлюкса, предотвращая патологическую перегрузку матрикса магнием и формируя устойчивый «катионный реостат».
3. Биоэнергетическое ядро: Специация нуклеотидов и контроль метаболической устойчивости
Биологическая активность АТФ неразрывно связана с магнием: в клеточной среде АТФ существует преимущественно в форме комплекса MgATP. Любое отклонение в уровне свободного Mg2+ ведет к изменению «специации нуклеотидов», делая энергетический пул функционально инертным для киназных сетей.
Узел аденилаткиназы как сенсор гомеостаза Магний выступает критическим регулятором аденилаткиназного узла, который устанавливает равновесие между пулами АТФ, АДФ и АМФ. Изменение концентрации Mg2+ индуцирует структурную реорганизацию активного центра аденилаткиназы, оптимизируя геометрию переноса фосфорила. Это делает магний гейткепером всей системы аденилатной сигнализации, включая тонус путей AMPK и NAD+.
Регуляция митохондриального потока Магний контролирует скорость-лимитирующие ферменты цикла трикарбоновых кислот:
- Изоцитратдегидрогеназу (IDH3), где ионы металла необходимы для аллостерической активации.
- Альфа-кетоглутаратдегидрогеназу (OGDH), определяющую продукцию восстановительных эквивалентов (NADH). На финальном этапе синтеза энергии Mg2+ координирует каталитический цикл F1Fo-ATP-синтазы, а селективный переносчик SCaMC (SLC25A23) обеспечивает направленный транспорт именно MgATP в цитозоль. Таким образом, магний является биоэнергетическим чекпоинтом, определяющим КПД превращения химической энергии в работу.
4. Ось «Магний–Кальций–Митохондрии» в патогенезе почечного повреждения
В условиях ишемического или токсического (например, при воздействии цисплатина) стресса потеря магния инициирует катастрофический каскад, превращая митохондрию из генератора энергии в медиатор клеточной смерти.
Последовательность ионного коллапса:
- Десинхронизация катионного реостата: Снижение матриксного Mg2+ снимает ингибирующий контроль с митохондриального кальциевого унипортера (MCU).
- Кальциевая экспансия: MRS2 и MCU начинают работать в режиме неконтролируемого входа Ca2+, провоцируя кальциевую перегрузку матрикса.
- Открытие mPTP и коллапс ΔΨm: Избыток кальция активирует открытие поры митохондриальной проницаемости (mPTP), что ведет к полной деполяризации мембраны.
- Генерация ROS и некроптоз: Ускоренная генерация активных форм кислорода (ROS) завершает гибель клетки.
Этот процесс является фундаментом «AKI-to-CKD перехода», где потеря магниевого буфера запирает канальцевый эпителий в состоянии метаболической ригидности и хронического воспаления.
5. Метаболическая негибкость и инсулинорезистентность: Почки как «метаболический амплификатор»
Дефицит магния деградирует верность инсулинового сигнала, формируя самоподдерживающуюся петлю обратной связи.
- Молекулярные механизмы резистентности: Низкий уровень MgATP ограничивает тирозинкиназную активность рецептора инсулина, ослабляя фосфорилирование IRS и активацию AKT. В отсутствие адекватного магниевого фона сигнальная сеть становится «дырявой», что блокирует транслокацию GLUT4.
- Роль стресс-киназ JNK и p38: Окислительный стресс, вызванный дефицитом Mg2+, активирует киназы JNK и p38. Эти медиаторы фосфорилируют IRS по ингибирующим сериновым остаткам, окончательно блокируя инсулиновый путь.
- Порочный круг: Гипергликемия через осмотический диурез усиливает почечные потери Mg2+, а гипомагниемия, в свою очередь, усугубляет инсулинорезистентность. Почки здесь функционируют как «амплификатор», ускоряя системную метаболическую декомпенсацию.
6. Гипотеза «Магниевых часов» (Magnesium Clock) и клеточное старение
Магний — это «темпоральный метаболит», чьи осцилляции регулируют биологическое время клетки.
Циркадная декомпозиция и энергетический шум В норме уровень Mg2+ совершает циркадные колебания, синхронизируя энергетический баланс. При старении наблюдается демпфирование амплитуды этих осцилляций. Переход от ритмической динамики к «энергетическому шуму» снижает порог устойчивости клетки к стрессу.
Триггеры сенильности и SASP Снижение митохондриального Mg2+ служит сигналом для активации путей p53 и p16, стабилизирующих необратимую остановку клеточного цикла. Логическая цепь инфламмэйджинга:
Дефицит Mg2+ → Дисфункция «катионного реостата» → Активация SASP → Системное воспаление → Почечная экскреция Mg2+ → Ускорение дряхлости (frailty).
Магний выступает «буфером буферов», и его истощение является скрытым темпоральным регулятором наступления клеточного старения.
7. Терапевтические перспективы: От восполнения к прецизионной модуляции
Современная стратегия требует перехода от неспецифического приема магния к компартмент-специфической модуляции биоэнергетики. supplementation в неселективных популяциях часто демонстрирует отсутствие эффекта; успех возможен только при механистической стратификации групп с доказанным дефицитом.
Трехуровневая модель интервенции (Tier 1-3):
- Идентификация: Анализ сывороточного магния лишь как «прокси-индикатора», дополненный оценкой почечного риска и медикаментозного фона.
- Динамическая верификация: Оценка гликемического и метаболического ответа на пробное восполнение депо.
- Таргетная коррекция: Устранение корневых причин (канальцевые потери) и разработка стратегий воздействия на баланс MRS2/SLC41A3 для сохранения митохондриальной целостности.
Итоговый вывод: Восстановление магниевого гомеостаза — это не просто нутритивная поддержка, а фундаментальная метаболическая интервенция. Она направлена на предотвращение «магниевого дрейфа» и сохранение здоровья почек как ключевого звена в продлении периода здоровой жизни (healthspan). Необходимы долгосрочные исследования in vivo, чтобы подтвердить потенциал сохранения MRS2-зависимого пула Mg2+ как инструмента замедления системного старения.