В современной инновационной дерматологии происходит фундаментальный сдвиг парадигмы: от упрощенной модели линейного воспаления мы переходим к концепции, где центральным патогенетическим драйвером выступает иммуностарение Т-лимфоцитов. Этот процесс не является синонимом пассивного угасания функции; напротив, это активная молекулярная трансформация, превращающая лимфоциты в автономные метаболические «фабрики» воспаления. Стратегическая значимость этого феномена заключается в формировании устойчивого к апоптозу воспалительного резервуара, который поддерживает хронизацию дерматозов даже при адекватной стандартной терапии.
1. Концепция Т-клеточного иммуностарения в современной дерматологии
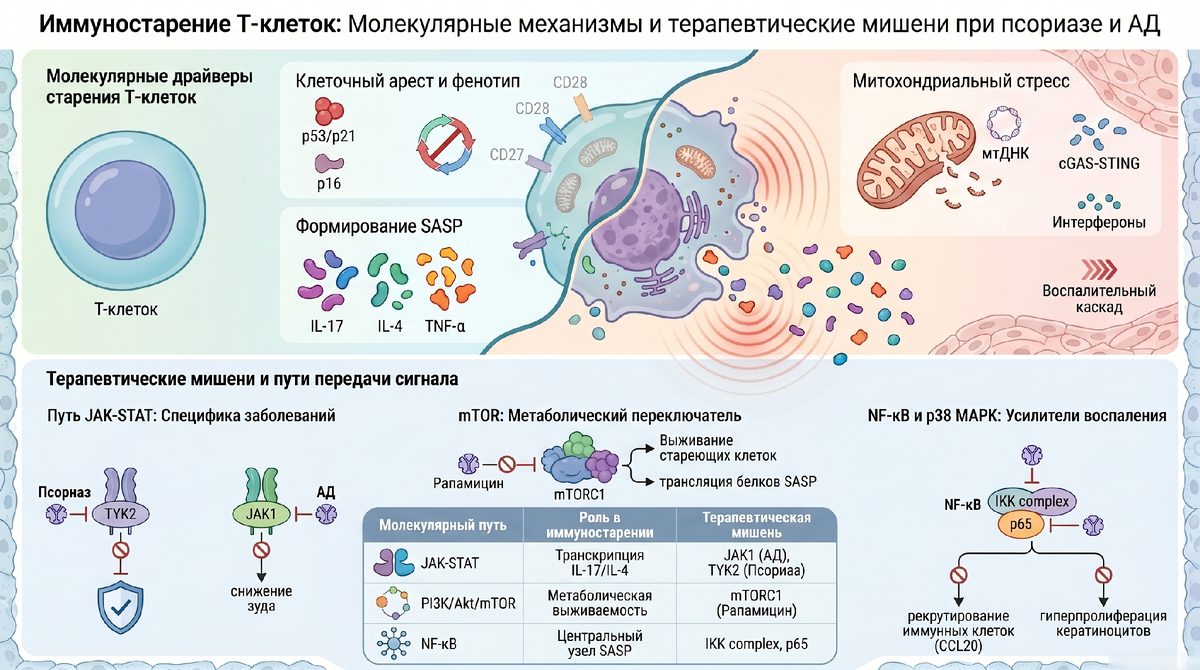
Т-клеточное иммуностарение определяется как прогрессирующий функциональный упадок, характеризующийся необратимым арестом клеточного цикла в G1-фазе при сохранении высокой метаболической и секреторной активности. В основе процесса лежат эпигенетические модификации комплекса TSC1/TSC2, митохондриальная дисфункция и формирование сенесцентно-ассоциированного секреторного фенотипа (SASP).
Критически важно проводить дифференциальную диагностику между «стареющими» и «истощенными» (exhausted) клетками, так как их терапевтический таргетинг требует диаметрально противоположных стратегий:
- Функциональный статус: Стареющие клетки демонстрируют метаболическую гиперреактивность, в то время как истощенные клетки находятся в состоянии глубокого функционального угнетения.
- Секреторный профиль: Стареющие Т-клетки являются первичными продуцентами SASP (IL-1, IL-6, TNF-α, MMPs), активно модулируя микроокружение.
- Драйверы и регуляторы: Старение управляется внутренними повреждениями (теломерная аттриция, оксидативный стресс), тогда как истощение — внешним избытком антигенной стимуляции и экспрессией PD-1/Tim-3.
- Резистентность: Стареющие клетки обладают аномальной живучестью за счет ап-регуляции антиапоптотических путей (BCL-2/BCL-xL), формируя постоянный источник патогенных сигналов.
Эти фенотипические сдвиги подготавливают почву для глубокой молекулярной перестройки сигнальных путей, делая клетку невосприимчивой к стандартным механизмам иммунного контроля.
2. Молекулярный профиль и фенотипические маркеры стареющих Т-клеток
Точная идентификация маркеров старения является необходимым условием для перехода от системной иммуносупрессии к селективной элиминации патогенных клонов. Накопление данных маркеров в дерме напрямую коррелирует с тяжестью псориаза и атопического дерматита, особенно у пациентов с длительным анамнезом заболевания (≥ 15 лет).
Биомаркерный анализ иерархии старения
- Внутриклеточные медиаторы ареста:p53: Мастер-регулятор, активируемый через ATM/ATR-каскад в ответ на геномную нестабильность.
p21 (CIP1): Прямой эффектор p53, ингибирующий комплекс Cyclin E/CDK2 и блокирующий фосфорилирование белка ретинобластомы (Rb).
p16 (INK4a): Специфический ингибитор CDK4/6, обеспечивающий необратимость G1-ареста и долгосрочное поддержание сенесцентного статуса. - Поверхностные маркеры:Утрата CD27/CD28: Свидетельствует о дефекте костимуляции и приобретении цитотоксического фенотипа.
Экспрессия CD57 и KLRG-1: Терминальные маркеры репликативного старения, коррелирующие с укорочением теломер. - Метаболические индикаторы:ROS (Активные формы кислорода): Продукт митохондриального стресса, повреждающий теломеры.
мтДНК (митохондриальная ДНК): Утечка мтДНК в цитозоль через поры BAK/BAX выступает основным эндогенным лигандом.
Патогенетический цикл cGAS-STING
Центральным механизмом формирования «порочного круга» воспаления является активация пути cGAS-STING. Распознавая цитозольную мтДНК, этот путь через TBK1 и IRF3 инициирует синтез интерферонов I типа. В синергии с NF-κB это приводит к массивному выбросу SASP-компонентов (CCL20, CXCL8, MMPs), которые рекрутируют новые воспалительные клетки, создавая устойчивый очаг тканевого повреждения.
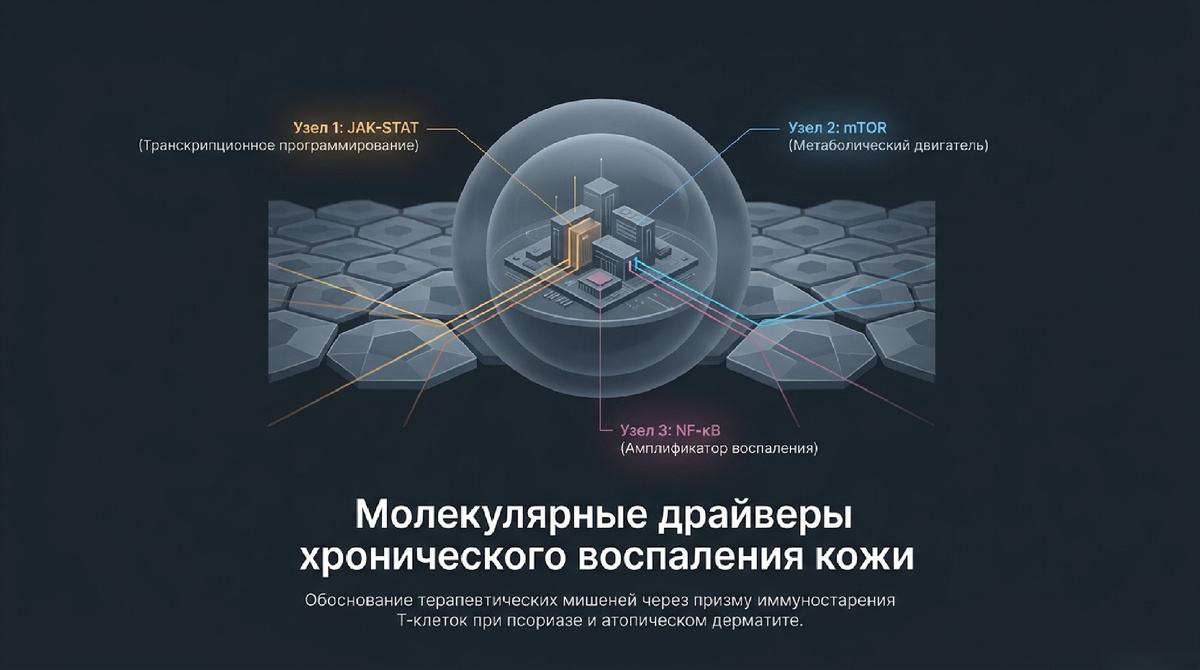
3. Анализ центральных сигнальных путей: JAK-STAT, mTOR и NF-κB
Сигнальные сети стареющей клетки организованы в жесткую иерархию, где JAK-STAT, mTOR и NF-κB выступают в роли интегративных «командных центров».
- Путь JAK-STAT: Гиперстимуляция субъединиц JAK2/TYK2 в стареющих лимфоцитах приводит к конститутивной активации STAT3. В псориатическом микроокружении это не только поддерживает синтез IL-17A, но и через транскрипцию IL-22 вызывает гиперпролиферацию кератиноцитов и акантоз.
- Путь PI3K-Akt-mTOR: mTORC1 функционирует как критический метаболический переключатель. Его активация блокирует аутофагию, что ведет к накоплению белка p62. Это, в свою очередь, через стабилизацию Nrf2 и NF-κB усиливает продукцию SASP и подавляет апоптоз. Постоянная активация mTORC1/S6K1 делает синтез воспалительных белков независимым от внешних ростовых факторов.
- Путь NF-κB и p38 MAPK: Ключевым молекулярным нюансом является фосфорилирование RelA по сайту S536, осуществляемое p38 MAPK. Этот процесс протекает независимо от деградации IκBα и многократно усиливает транскрипционную активность NF-κB. Результатом становится синергическое усиление инфильтрации тканей и стабилизация мРНК провоспалительных цитокинов.
4. Патогенетическая сегрегация: Псориаз vs Атопический дерматит (AD)
Несмотря на общность механизмов старения, молекулярная архитектура ответа диктуется специфичностью иммунных осей Th17 и Th2.
- Псориаз: Доминирует ось IL-23/JAK2-TYK2/Th17. Стареющие клетки через продукцию CCL20 формируют хемотаксический градиент для рекрутирования CCR6+ патогенных лимфоцитов. Особую роль играет cGAS-STING-индуцированный IFN-α, придающий псориазу черты аутовоспалительного процесса.
- Атопический дерматит: Основным драйвером является каскад JAK1/STAT6, активируемый IL-4 и IL-13. В стареющих Т-клетках этот путь подавляет экспрессию филаггрина и лорикрина через ингибирование транскрипционных факторов Ovol-1/Grhl3. Хронический зуд поддерживается через гиперпродукцию IL-31, замыкающего цикл «зуд-расчесы-воспаление».
- Розацеа и себорейный дерматит: При розацеа критическим фактором является возрастное снижение SIRT7, что активирует ось TLR2-NF-κB и продукцию LL37. При себорейном дерматите метаболиты Malassezia и свободные fatty acids (FFA) активируют NF-κB, создавая липидно-воспалительный цикл.
5. Стратегии таргетной терапии и клинический статус (март 2026)
Стратегический вектор смещается от купирования симптомов (ингибирование SASP) к прецизионной модуляции апстрим-регуляторов иммуностарения.
Аналитический обзор ингибиторов Янус-киназ (JAKi)
Препарат | Селективность | Основное показание | Статус (март 2026) | Клинический комментарий
Icotrokinra (JNJ-77242113) | Рецептор IL-23R | Псориаз | Одобрен (FDA) | Превосходит Deucravacitinib по частоте PASI-90 за счет прямого блока p19-оси.
Deucravacitinib | TYK2 (аллостерический) | Псориаз | Одобрен | Золотой стандарт таргетной терапии Th17-зависимых дерматозов.
Upadacitinib | JAK1 | AD, Псориат. артрит | Одобрен | Эффективен при резистентности к Дупилумабу за счет широкого блока IL-4/13/22/31.
Abrocitinib | JAK1 | AD | Одобрен | Демонстрирует превосходство над биологиками в купировании зуда (NRS) на 1-й неделе.
Zasocitinib | TYK2 | Псориаз | Фаза II (Завершено) | Высокая селективность, минимальные системные риски.
Delgocitinib | JAK1/2/3, TYK2 | Хроническая экзема | Одобрен | Топический пан-JAK ингибитор с высокой локальной эффективностью.
Прецизионные биологические агенты
- Модуляторы IL-4Rα: Stapokibart (CM310) демонстрирует более высокие показатели EASI-75 по сравнению с Дупилумабом. Это обусловлено его уникальным эпитопом связывания, расположенным ближе к лиганд-связывающему центру, что обеспечивает более мощную блокировку Th2-SASP.
- Анти-IL-31: Nemolizumab остается лидером по скорости наступления эффекта (улучшение сна и NRS на 1-й неделе), опережая лебрикизумаб и тралокинумаб.
- Анти-IL-23/17: Tildrakizumab выделяется способностью модулировать метилирование ДНК, потенциально замедляя «эпигенетические часы» иммунных клеток.
6. Вызовы и горизонты: прямой таргетинг сенесцентных клеток
Главным вызовом остается «воспалительный резервуар» — стареющие Т-клетки, которые не элиминируются текущими биологиками. Это обуславливает высокую частоту рецидивов после отмены терапии.
Перспективы сенотерапии:
- Сенолитики: Использование ингибиторов BCL-2 (ABT-737) позволяет преодолеть апоптотическую резистентность стареющих клеток. Экспериментальные данные 2025-2026 гг. подтверждают, что топическое применение ABT-737 нормализует TCR-репертуар и подавляет Th17-SASP.
- Метаболическое омоложение: Агонисты AhR (Tapinarof) и восстановление пула NAD+ (через NMN) позволяют восстановить активность SIRT1/SIRT7, что подавляет ацетилирование p65 и STAT3, возвращая клетку под эпигенетический контроль.
- Диетарные стратегии: Интервальное голодание и модуляция кофеином показали способность снижать популяцию CD57+ Т-клеток в дерме, воздействуя на mTOR-зависимые механизмы старения.
Заключение: Будущее дерматологии — в достижении «глубокой ремиссии» через таргетную элиминацию сенесцентных клонов. Переход от нейтрализации цитокинов к управлению биологическим возрастом иммунной системы (например, через модуляцию метилирования ДНК Тильдракизумабом) открывает путь к истинному излечению хронических дерматозов.