1. Введение: Старение как детерминанта сердечной недостаточности
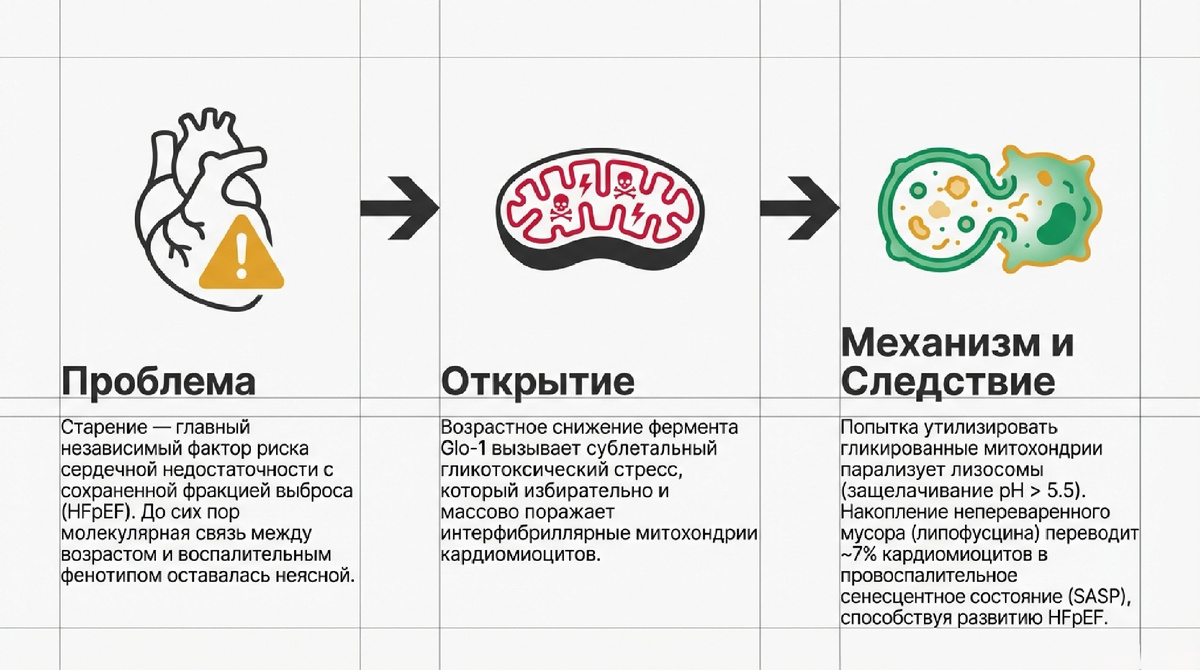
Понимание механизмов клеточного старения имеет фундаментальное значение для современной кардиологии, особенно в контексте патофизиологии сердечной недостаточности с сохраненной фракцией выброса (HFpEF). Кардиомиоциты, представляя собой долгоживущие постмитотические клетки, лишены возможности снижать концентрацию поврежденных макромолекул путем клеточного деления. В условиях исключительно высокой интенсивности окислительного метаболизма миокарда стохастическая химическая атака побочных продуктов обмена на белковые структуры приводит к экспоненциальному накоплению повреждений. Ключевым фактором этого процесса выступает «гликирующий стресс» (glycative stress), провоцирующий прогрессирующую потерю молекулярной точности (molecular fidelity) белков. В основе данного состояния лежит коллапс систем детоксикации реактивных дикарбонилов, что превращает кардиомиоцит в резервуар токсичных аддуктов, инициирующих каскад возрастной дегенерации ткани.
2. Гликирующий стресс и метаболический коллапс системы глиоксалаз
Дикарбонильный стресс в миокарде является следствием накопления реактивных α-оксоальдегидов, возникающих как побочные продукты гликолиза. Высокая химическая активность этих соединений создает условия для необратимой модификации протеома.
- Роль α-оксоальдегидов: Метилглиоксаль (MGO) и глиоксаль (GO) выступают основными медиаторами гликирования. Обладая высоким сродством к положительно заряженным аминокислотным остаткам, они нарушают растворимость и функциональную конформацию белков.
- Система детоксикации: Универсальным механизмом защиты является система глиоксалаз (Glo-1 и Glo-2), где критическим ограничивающим фактором выступает глутатион (GSH) в качестве кофактора. Согласно данным исследований, старение (≥20 месяцев у мышей и ≥75 лет у людей) сопровождается сочетанным дефицитом активности Glo-1 и уровня глутатиона. Этот протеостатический коллапс делает спонтанное образование аддуктов термодинамически неизбежным.
- Классификация конечных продуктов гликирования (AGEs): В стареющем миокарде идентифицирована гетерогенная группа устойчивых к протеолизу соединений, среди которых наиболее патофизиологически значимы:Arg1HPG и Arg2HPG (производные аргинина, маркеры выраженного старения);
MG-H1 (MGO-производный гидроимидазолон-1);
CML (карбоксиметиллизин);
MDA54 (лизиновый димер);
G-H1 (глиоксаль-производный аддукт).
Снижение эффективности Glo-зависимой утилизации превращает внутриклеточные компартменты, прежде всего митохондрии, в основные депо AGEs.
3. Митохондрии как первичная мишень гликирования: селективность поражения
Поддержание митохондриального протеостаза является императивом для энергетического гомеостаза сердца. Однако масс-спектрометрический анализ подтверждает избирательную уязвимость органелл: до 26% всех внутриклеточных AGEs локализовано именно в митохондриях. Критической молекулярной мишенью гликирующего повреждения выступает FoF1-ATP синтаза, модификация которой напрямую лимитирует синтез АТФ.
Старение миокарда характеризуется выраженной селективностью поражения различных субпопуляций митохондрий:
Параметр сравнения / Интерфибриллярные митохондрии (IFM) / Субсарколеммальные митохондрии (SSM)
Уровень накопления AGEs / Значительное повышение (MAGE, CML, Arg1/2HPG) / Стабильный уровень, отсутствие накопления
Потребление O2 (State 3) / Существенное снижение (комплекс-1 зависимое) / Сохранено на функциональном уровне
Дыхательный контроль (RCR) / Снижен (дыхательное разобщение) / Сохранен
Морфологические изменения / Аберрантная организация крист («луковичная» морфология), гигантизм / Преимущественная сохранность ультраструктуры
Селективное поражение IFM, обеспечивающих энергией сократительный аппарат, фатально снижает адаптационный резерв сердца к нагрузкам, что формирует субстрат для развития HFpEF даже при нормальных показателях фракции выброса в покое.
4. Дисфункция митохондриально-лизосомальной оси и накопление липофусцина
В норме контроль качества органелл осуществляется через митофагию, однако гликирующий стресс разрывает функциональную связь между митохондриями и лизосомами.
- Нарушение лизосомального гомеостаза: В стареющих кардиомиоцитах наблюдается выраженное защелачивание лизосом (падение соотношения LSG/LTR). Первичной причиной этого процесса является прямое воздействие AGE-модифицированных митохондрий, которые при попытке захвата подавляют протонную помпу лизосом.
- Механизм незавершенного стресса: Гликированные белки крайне резистентны к гидролизу. Это ведет к накоплению митохондриальных везикул (MDVs) и фрагментов, которые не могут быть эффективно утилизированы, формируя порочный круг органеллярной дисфункции.
- Генез липофусцина: Липофусцин — агрегат непереваренного материала, обогащенный AGEs и продуктами окисления белков. Его накопление в форме электроплотных гранул служит морфологическим маркером «незавершенного» аутофагического ответа.
- Фенотип гигантских митохондрий: Неспособность лизосом к поглощению крупных объектов приводит к сохранению в цитоплазме гиперразветвленных, «луковичных» митохондриальных структур, которые продолжают генерировать активные формы кислорода при минимальном выходе АТФ.
5. Героконверсия кардиомиоцитов и формирование SASP
Хроническое накопление непереваренного материала и дисфункциональных органелл инициирует программу героконверсии — перехода клетки в сенесцентное состояние.
- Биомаркеры и распространенность: Ключевыми индикаторами являются повышение активности SA-β-Gal и экспрессия p16INK4a и p15INK4b (при стабильном уровне p21CIP1). Установлено, что около 7% кардиомиоцитов в стареющем сердце приобретают сенесцентный фенотип.
- Скрытая патология: Важно подчеркнуть, что данный фенотип развивается в клинически асимптомном миокарде. Однако паракринного влияния этих 7% клеток достаточно для радикальной трансформации тканевой микросреды через секреторный фенотип (SASP).
- Структура SASP:Провоспалительные медиаторы: IL-1a, IL-6;
Хемокины: Ccl2, Cxcl1 (стимуляция инфильтрации иммунными клетками);
Факторы ремоделирования: Mmp9, TGF-β2 (стимуляция фиброза и жесткости матрикса).
Секреция этих факторов превращает миокард в хроническую воспалительную среду, характерную для HFpEF, предопределяя переход возрастных изменений в клиническую стадию болезни.
6. Заключение: Синтез данных и терапевтические перспективы
Представленные данные позволяют выстроить четкую патофизиологическую последовательность: возрастной дефицит системы глиоксалаз и глутатиона -> накопление AGEs на FoF1-ATP синтазе -> селективная деградация интерфибриллярных митохондрий -> лизосомальное защелачивание -> героконверсия кардиомиоцитов с развитием воспалительного SASP. Идентификация накопления митохондриальных AGEs как пускового механизма открывает новые горизонты для терапевтического воздействия. Перспективными стратегиями являются реактивация Glo-1 для превентивной детоксикации дикарбонилов и искусственное подкисление лизосом для восстановления митофагии, что может предотвратить развитие возрастной сердечной недостаточности.