Гипопаратиреоз - cостояние, характеризующееся сниженной продукцией паратиреоидного гормона (ПТГ) околощитовидными железами (ОЩЖ) или резистентностью тканей к его действию, что приводит к различным нарушениям, прежде всего фосфорно-кальциевого обмена с развитием гипокальциемии, гиперфосфатемии и гиперкальциурии. Гипопаратиреоз встречается у 0,2-0,3% населения, ~23-37случая на 100 000 населения. Послеоперационный гипопаратиреоз чаще встречается среди женщин, что связано с более частой патологией ЩЖ и, следовательно, тиреоидэктомией. Частота наследственных форм гипопаратиреоза не различается у мужчин и женщин.
Гипокальциемия - стойкое снижение содержания общего кальция в плазме крови менее 1,87 ммоль/л, а ионизированного кальция - менее 1,07 ммоль/л.
Основные механизмы развития гипокальциемии:
- Заболевания ОЩЖ, приводящие к гипопаратиреозу;
- Резистентность тканей-мишеней к ПТГ, псевдогипопаратиреоз;
- Дефицит витамина D или резистентность к нему, нарушение метаболизма витамина D;
- Недостаточное поступление кальция с пищей, нарушение всасывания кальция в кишечнике;
- Повышенное выведение кальция при почечной недостаточности и др.;
- Подавление синтеза и секреции ПТГ, в том числе лекарственными средствами;
- Гипомагниемия или гипермагниемия;
- Усиленный захват кальция костной тканью.
Заболевания и состояния, приводящие к развитию гипокальциемии
Недостаточность секреции паратгормона
- Послеоперационный гипопаратиреоз;
- Гипопаратиреоз после лечения радиоактивным йодом;
- Гипопаратиреоз, развившийся в результате травмы, саркоидоза, туберкулезного поражения ОЩЖ;
- Гипопаратиреоз вследствие первичной или вторичной опухоли шеи с разрушением ОЩЖ.
- Гипопаратиреоз аутоиммунного генеза, в том числе полиэндокринный аутоиммунный синдром, семейный гипопаратиреоз аутоиммунного генеза с недостаточностью многих эндокринных желез и кандидозом (МЕDАС – синдром); синдром Ди Джоржи – агенез ОЩЖ, аплазия вилочковой железы, врожденные уродства, иммунологическая недостаточность;
Функциональный гипопаратироз (недостаточная секреция ПТГ в ответ на гипокальциемию)
- Гипопаратиреоз новорожденных, родившихся от матерей, страдающих гипопаратирозом;
- Идиопатическая неонатальная гипокальциемия;
- Гипомагниемия (малабсорбция, рвота и диарея, стеаторея, сахарный диабет, острый панкреатит и др.);
- Недостаток витамина D (алиментарный дефицит, недостаток УФ-лучей, малабсорбция, стеаторея, синдром короткого кишечника, хронический панкреатит, спру и др.).
Периферическая резистентность к паратгормону
- Псевдогипопаратиреоз (синдром Олбрайта);
- Гипомагниемия;
- Хроническая почечная недостаточность;
- Дефицит витамина D.
Гипокальциемия ятрогенной природы
- Введение фосфатов (в том числе избыток фосфора в пище);
- Введение ЭДТА (этилендиаминтетрауксусная кислота);
- Введение митрамицина, актиномицина, неомицина;
- Применение тиазидовых диуретиков;
- Длительное применение слабительных;
- Длительный прием фенобарбитала и других противосудорожных средств;
- Массивная трансфузия цитратной крови, операция в условиях экстракорпорального кровообращения;
- Передозировка кальцитонина;
- Применение бифосфонатов.
Этиология гипопаратиреоза
Послеоперационный гипопаратиреоз – самый частый вариант гипопаратиреоза (до 63-91% случаев), связанный с непосредственным удалением, интраоперационной травмой или нарушением кровоснабжения ОЩЖ при хирургических операциях на органах шеи.
Аутоиммунный гипопаратиреоз – связан с аутоиммунным поражением ОЩЖ; встречается редко; может быть самостоятельным заболеванием (изолированный, первичный аутоиммунный гипопаратиреоз) или быть в составе аутоиммунного полиэндокринного синдрома с одновременным поражением коры надпочечников, инсулярного аппарата поджелудочной железы и поражение других эндокринных желез.
Генетический гипопаратиреоз (<10% случаев) – может быть изолированным или встречаться в рамках поликомпонентных генетических синдромов (синдром Ди Джорджи, синдром Бараката, синдром Саньяд-Сакати и др.).
Другие формы гипопаратиреоза: посттравматический (после травмы ЩЖ и ОЩЖ), воспалительный (при тиреоидитах), пострадиационный (после радиойодтерапии), метастатический и идиопатический гипопаратиреоз.
Классификация гипопаратиреоза по этиологии
Послеоперационный гипопаратиреоз:
- Транзиторный гипопаратиреоз;
- Хронический (стойкий) гипопаратиреоз.
Аутоиммунный гипопаратиреоз:
- Аутоиммунный полигландулярный синдром 1 типа (АПС 1 типа);
- Аутоиммунный полигландулярный синдром 3 типа (АПС 3 типа);
- Аутоиммунный полигландулярный синдром 4 типа (АПС 4 типа).
Генетический изолированный гипопаратиреоз:
- Аутосомно-доминантная гипокальциемия с гиперкальциурией 1 типа (HYPOC1-ADH1)/синдром Барттера 5 типа;
- Аутосомно-доминантная гипокальциемия с гиперкальциурией 2 типа (HYPOC1-ADH2);
- Семейный изолированный гипопаратиреоз (аутосомный, Х-связанный).
Гипопаратиреоз в составе поликомпонентных генетических синдромов:
- Синдром Ди Джорджи 1 типа (DGS1, Di George syndrome 1);
- Синдром Ди Джорджи 2 типа (DGS2, Di George syndrome 2);
- CHARGE синдром;
- Синдром Бараката (Barakat), HDR-синдром (hypoparathyroidism, deafness and renal dysplasia syndrome);
- Синдром Кенни-Каффи 1 типа (KCS1, Kenny Caffey 1);
- Синдром Кенни-Каффи 2 типа (KCS2, Kenny Caffey 2);
- Gracile bone dysplasia (GCLEB);
- Митохондриальные заболевания или болезни митохондриальной ДНК (синдром Кернса-Сейра, синдром MELAS (mitochondrial encephalopathy, lactic acidosis and stroke-like episodes), синдром MTPDS).
Другие формы гипопаратиреоза:
- Нарушения обмена магния;
- Инфильтративные заболевания (гранулематоз, гемохроматоз, метастазирование, амилоидоз и др.);
- Гипопаратиреоз в результате лучевого повреждения ткани ОЩЖ (радиойодтерапия при патологии ЩЖ).
Идиопатический гипопаратиреоз
Виды гипопаратиреоза
Приобретенный гипопаратиреоз: а) послеоперационный; б) аутоиммунный (изолированный); в) возникающий при разрушении ОЩЖ: в результате лучевого воздействия; вследствие инфильтративных заболеваний (гемосидероз, болезнь Вильсона-Коновалова, гранулематоз, амилоидоз).
Врожденный гипопаратиреоз: а) изолированный; б) в составе наследственных синдромов: Ди Джоржи; Кенни - Каффи; Бараката; аутоиммунного полигландулярного синдрома 1-го типа; в) ассоциированный с митохондриальными нарушениями (Кернса - Сейра синдром, MELAS, MTPDS).
Псевдогипопаратиреоз – редкое наследственное заболевание, характеризующееся резистентностью органов-мишеней к ПТГ и проявляющееся клинико-лабораторными признаками гипопаратиреоза на фоне повышенного уровня ПТГ в крови.
Послеоперационный гипопаратиреоз – при хирургических вмешательствах в области шеи.
- Транзиторный послеоперационный гипопаратиреоз – гипокальциемия, развивающаяся в первые 24 часа после хирургической операции на органах шеи и разрешающаяся в течение 4-6 месяцев.
- Хронический (стойкий) послеоперационный гипопаратиреоз – гипокальциемия в сочетании с низким уровнем ПТГ спустя 6 месяцев и более после хирургического операций на органах шеи.
Латентный (бессимптомный) гипопаратиреоз - протекает без клинических проявлений и выявляется только при лабораторном исследовании гипокальциемией и низким уровнем ПТГ.
Манифестный (симптомный) гипопаратиреоз – протекает с клиническими и лабораторными проявлениями гипопаратиреоза и может быть острым или хроническим.
Состояние фосфорно-кальциевого обмена при гипопаратиреозе
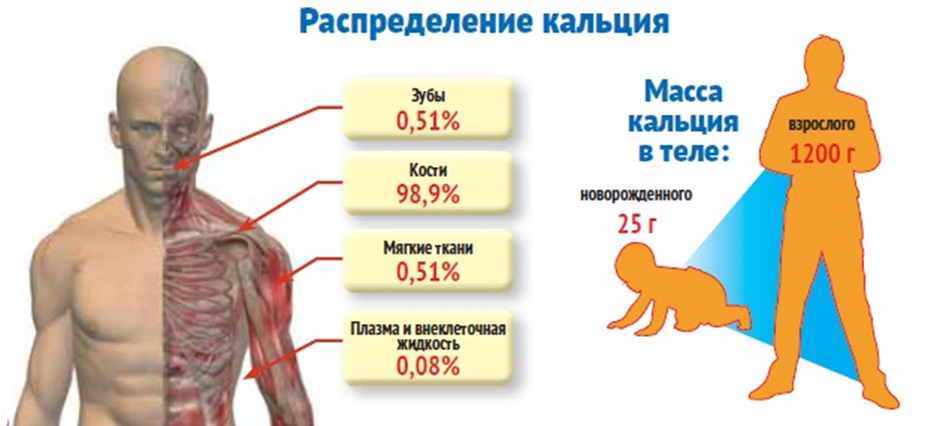
Более 90% кальция депонировано в костях, преимущественно в виде гидроксиапатита кальция. Во внеклеточной жидкости содержится менее 1% кальция, а его уровень внутри клеток чрезвычайно низок.
Кальций во внеклеточной жидкости присутствует в 3 формах: связанный с белками, в основном, с альбумином (около 40%) и не фильтруемый почками; связанный с органическими анионами (около 10%); активная свободная ионизированная форма кальция, определяющая его физиологическое действие, связываясь с кальций-чувствительным рецептором (calcium-sensing receptor, CaSR).
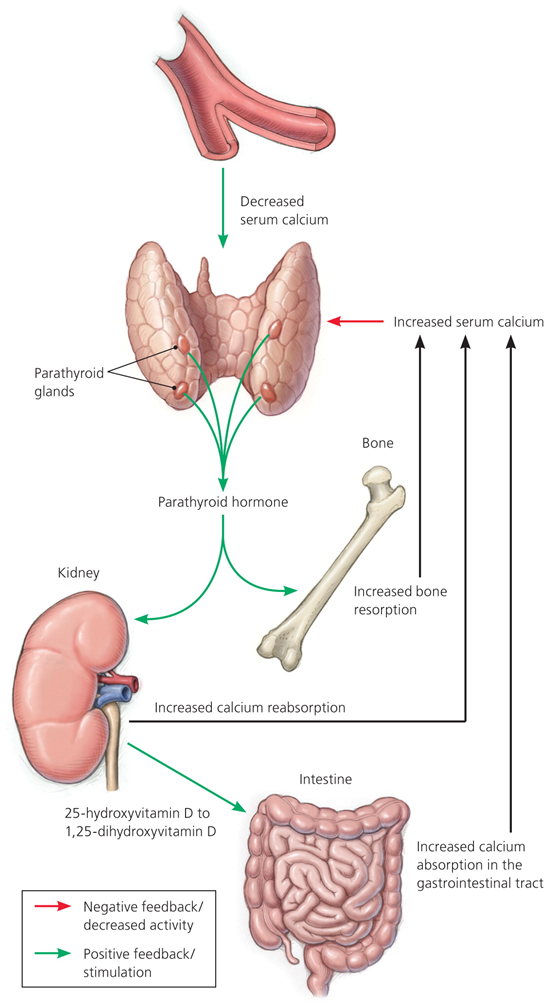
ПТГ – основной регулятор фосфорно-кальциевого обмена, синтезируется и секретируется главными клетками ОЩЖ в ответ на снижение уровня кальция сыворотки крови, определяемое CaSR, расположенными на поверхности паратиреоцитов. Гипокальциемия – основной фактор, влияющий на синтез и секрецию ПТГ. К другим факторам, влияющим на продукцию ПТГ относятся витамин D, фосфор, магний, а также фактор роста фибробластов 23 (ФРФ23), эффекты которого заключаются в подавлении активности 1α-гидроксилазы в почках, снижении реабсорбции фосфора и секреции ПТГ.
В почках ПТГ повышает реабсорбцию кальция в толстой части восходящей петли Генле и дистальных извитых канальцах, снижает реабсорбцию фосфора в проксимальных канальцах.
В проксимальных канальцах почек ПТГ стимулирует 1α-гидроксилазу, которая ответственна за образование активной формы витамина D - 1,25-дигидроксихолекальциферола (кальцитриола).
Кальцитриол главным образом повышает всасывание кальция и фосфора в кишечнике.
В костях ПТГ связывается с рецепторами ПТГ 1 типа (PTHR1) на остеобластах и остеоцитах.
Под воздействием ПТГ остеобласты вырабатывают медиаторы, в том числе фактор дифференцировки остеокластов (RANKL), который связывается с рецептором активатора нуклеарного фактора κB (RANK).
Активация гена транскрипции остеокластов и дифференцировка преостеокластов в зрелый остеокласт повышает костную резорбцию и высвобождение кальция и фосфора в кровоток.
Кроме того, кальцитриол усиливает экспрессию RANKL, повышает количество и активность остеокластов и стимулирует костную резорбцию, что приводит к повышению уровня кальция и фосфора в крови.
Также кальцитриол стимулирует экспрессию ФРФ23 остеоцитами в костях, что ингибирует секрецию ПТГ и почечную 1α-гидроксилазу и увеличивает экскрецию фосфатов почками.
Повышение уровня ионизированного кальция крови снижает синтез и секрецию ПТГ, снижает активность 1α-гидроксилазы, стимулирует парафолликулярные клетки ЩЖ, что приводит к высвобождению кальцитонина и подавлению костной резорбции, повышению экскреции кальция почками.
В результате описанных процессов достигается и поддерживается нормальный уровень кальция сыворотки крови.
При гипопаратиреозе отсутствие или недостаточность ПТГ сопровождается развитием гипокальциемии.
Основные патогенетические механизмы развития гипокальциемии при дефиците ПТГ:
- Снижение активности остеокластов с уменьшением высвобождения кальция из костей;
- Повышение экскреции кальция с мочой;
- Подавление синтеза кальцитриола в почках;
- Снижение абсорбции кальция из кишечника.
Дефицит ПТГ приводит к гиперфосфатемии как напрямую посредством увеличения почечной тубулярной реабсорбции фосфатов, так и косвенно за счет гипокальциемии.
Хроническая гиперфосфатемия у пациентов с гипопаратиреозом ассоциирована с повышением в крови уровня фактора роста фибробластов 23 (ФРФ23).
Отрицательный кальциевый баланс и избыток фосфора ведет к повышению нервно-мышечной возбудимости и общей вегетативной реактивности с повышением судорожной активности.
Магний также способен регулировать секрецию ПТГ аналогично кальцию: • Гипермагниемия активирует CaSR в ОЩЖ, подавляя секрецию ПТГ, и в дистальных почечных канальцах, снижая реабсорбцию кальция и магния; • Длительная гипомагниемия также приводит к развитию гипокальциемии, т.к. истощает внутриклеточные запасы магния, что приводит к активации CaSR, блокаде секреции ПТГ и, в конечном счете, вторичной гипокальциемии из-за снижения реабсорбции кальция почками.
Послеоперационный гипопаратиреоз
Послеоперационный гипопаратиреоз развивается после хирургических вмешательств на органах шеи (тиреоидэктомия и др.), составляет до 75% и более всех случаев гипопаратиреоза и может быть обусловлен как непосредственным удалением, так и интраоперационной травмой или нарушением кровоснабжения ОЩЖ.
К основным факторам риска развития послеоперационного гипопаратиреоза относят: большой объем хирургического вмешательства (как правило, по поводу рака ЩЖ, особенно с центральной лимфодиссекцией, диффузного токсического зоба), повторные вмешательства на органах шеи, а также ревизионные вмешательства.
К дополнительным факторам риска развития послеоперационного гипопаратиреоза относят: женский пол; дефицит витамина D; беременность и лактацию; аутоиммунные заболевания ЩЖ; мальабсорбция (например, после гастрошунтирования (бариатрическая хирургия), при воспалительных заболеваниях кишечника).
Транзиторный послеоперационный гипопаратиреоз определяется как снижение уровня ПТГ в сочетании с гипокальциемией в течение не более чем 6 месяцев после операции и регистрируется у 25-30% пациентов, перенесших тотальную тиреоидэктомию. Около 60-70% случаев послеоперационной гипокальциемии имеют транзиторный характер и разрешаются в течение 4-6 недель после операции.
Хронический послеоперационный гипопаратиреоз определяется при сохранении стойкой гипокальциемии в сочетании с низким или низко нормальным уровнем ПТГ спустя 6 и более месяцев после хирургического вмешательства в области шеи. Риск хронического гипопаратиреоза тесно связан с количеством оставшихся функционирующих ОЩЖ во время операции: 16% при сохраненных 1-2 железах, 6% при 3 железах и 2,5% при 4 железах. В редких случаях послеоперационный гипопаратиреоз может развиться спустя годы после операции, механизм чего до конца не изучен, но возможно связан с возрастными изменениями сосудистой стенки артерий, кровоснабжающих остаточную ткань ОЩЖ.
Генетические причины гипопаратиреоза
Генетическая этиология гипопаратиреоза встречается менее чем в 10% случаев. Основную причину гипопаратиреоза представляют собой хромосомные микроделеции и моногенные аномалии.
К генетически обусловленным вариантам гипопаратиреоза относят:
- Аутоиммунный гипопаратиреоз:
- Аутоиммунный полигландулярный синдром 1 типа (АПС 1 типа);
- Аутоиммунный полигландулярный синдром 3 типа (АПС 3 типа);
- Аутоиммунный полигландулярный синдром 4 типа (АПС 4 типа).
Генетический изолированный гипопаратиреоз:
- Аутосомно-доминантная гипокальциемия с гиперкальциурией 1 типа (HYPOC1-ADH1)/синдром Барттера 5 типа;
- Аутосомно-доминантная гипокальциемия с гиперкальциурией 2 типа (HYPOC1-ADH2);
- Семейный изолированный гипопаратиреоз (аутосомный, Х-связанный).
Гипопаратиреоз в составе поликомпонентных генетических синдромов:
- Синдром Ди Джорджи 1 типа (DGS1);
- Синдром Ди Джорджи 2 типа (DGS2);
- CHARGE синдром;
- Синдром Бараката, HDR-синдром (hypoparathyroidism, deafness and renal dysplasia syndrome);
- Синдром Кенни-Каффи 1 типа (KCS1);
- Синдром Кенни-Каффи 2 типа (KCS2);
- Gracile bone dysplasia (GCLEB);
- Митохондриальные заболевания или болезни митохондриальной дезоксирибонуклеиновой кислоты (синдром Кернса-Сейра, синдром MELAS (mitochondrial encephalopathy, lactic acidosis and stroke-like episodes), синдром MTPDS).
Аутоиммунный гипопаратиреоз
Аутоиммунный гипопаратиреоз – вторая по распространенности форма гипопаратиреоза, обусловленная иммуно-опосредованным разрушением клеток ОЩЖ. Он может быть изолированным заболеванием, однако чаще встречается в рамках наследственного аутоиммунного полигландулярного синдрома 1 типа (АПС 1 типа, кандидо-эктодермальная дистрофия, Autoimmune Polyendocrinopathy with Candidiasis and Ectodermal Dystrophy – APECED). Распространенность АПС 1 типа составляет от 1:9000-25000 до 1:100 000 населения. АПС 1 типа – моногенное аутосомно-рецессивное заболевание, в основе которого лежит нарушение структуры гена аутоиммунного регулятора (AIRE), располагающегося на длинном плече 21-й хромосомы, кодирующего ядерный фактор транскрипции и играет одну из ключевых ролей в формировании иммунотолерантности. В основе патогенеза АПС 1 типа лежит аутоиммунная деструкция различных эндокринных желез, включая ОЩЖ.
Главные компоненты АПС 1 типа: слизисто-кожный кандидоз, гипопаратиреоз, первичная хроническая надпочечниковая недостаточность. Другие возможные компоненты АПС 1 типа: сахарный диабет 1 типа; аутоиммунный гепатит; тиреоидит Хашимото; полиартрит; кератит; ретинит; первичная овариальная дисфункция; гипоплазия зубной эмали; энтеропатия с хронической диареей или запорами; фотофобия; периодический жар с сыпью; пневмонит; нефрит; панкреатит; функциональная аспления; целиакия; аплазия красного ростка костного мозга; целиакия; витилиго; гипогонадизм. Заболевание дебютирует, как правило, в детском возрасте. В подавляющем большинстве случаев первым проявлением становится слизисто-кожный кандидоз, развивающийся в первые 10 лет жизни, чаще в возрасте около 2 лет. На фоне слизисто-кожного кандидоза у 84% пациентов появляется гипопаратиреоз, при этом в 88% случаев он дебютирует в возрасте до 10 лет. У 30% пациентов находят аутоантитела к ПТГ.
Генетический изолированный гипопаратиреоз
В зависимости от патогенеза генетические варианты изолированного гипопаратиреоза условно могут быть разделены на три основные группы:
- Заболевания, характеризующиеся нормальным развитием ОЩЖ, но нарушенной секрецией ПТГ. К данной группе относится аутосомно-доминантная гипокальциемия, в основе которой лежат активирующие точечные мутации гена кальций-чувствительного рецептора CASR. Характерна умеренная гипокальциемия в сочетании со снижением уровня ПТГ, у большинства пациентов наблюдается гипомагниемия. Клиника может быть от бессимптомной гипокальциемии до гипокальциемических кризов. Характерна гиперкальциурия с высоким риском нефролитиаза/нефрокальциноза и нарушения функции почек.
- Аномалии развития ОЩЖ, возникающие в результате мутации транскрипционных факторов. Дефект гена GCM2 (glial cell missing 2) лежит в основе нарушения формирования ОЩЖ и наследуется по аутосомно-рецессивному типу. X-сцепленный рецессивный вариант связывают с делециями и инсерциями на участке Xq27.1.
- Мутации гена ПТГ, приводящие к нарушению процессинга и секреции ПТГ, сопровождающиеся развитием изолированного гипопаратиреоза. Данные мутации встречаются достаточно редко, имеют аутосомно-рецессивный и аутосомно-доминантный типы наследования. Нарушается посттрансляционная обработка молекулы препроПТГ и/или трансляции матричной рибонуклеиновой кислоты. Гомозиготные мутации гена препроПТГ приводят к значимому снижению уровня ПТГ (вплоть до неопределяемых значений) в сочетании с симптоматической гипокальциемией и гиперфосфатемией.
Гипопаратиреоз в составе поликомпонентных генетических заболеваний
Гипопаратиреоз может входить в состав поликомпонентных генетических заболеваний.
Синдром Ди Джорджи (Di George syndrome, синдром Ди Джорджа, синдром Ди Георга, синдром делеции 22q11.2) является следствием неправильного развития органов-производных третьего и четвертого жаберных карманов. Заболевание встречается у 1 из 4000-5000 новорожденных, и в большинстве случаев (70-80%) является следствием гетерозиготной микроделеции в 22q11.21-q11.23 участках, что приводит к врожденной дисгенезии ОЩЖ и соответственно к гипопаратиреозу. Большинство случаев являются спорадическими, но встречаются семейные случаи с аутосомно-доминантным типом наследования синдрома. Больные обычно погибают в детском возрасте. Клинически синдром Ди Джорджи проявляется задержкой в развитии, «волчьим небом», аплазией или гипоплазией ОЩЖ и тимуса, врожденными пороками сердца, характерным внешним видом (квадратный корень носа) и тяжелым иммунодефицитом. Синдром Ди Джорджи – ведущая причина персистирующей гипокальциемии у новорожденных, но гипопаратиреоз может проявиться как в неонатальном периоде, так и позднее в течение жизни. Среди всех людей с этим заболеванием гипопаратиреоз встречается у 60% пациентов.
Спектр клинических проявлений при синдроме Ди Джорджи
- Врожденные пороки сердца представлены не менее, чем в 80% случаев (чаще прерывание дуги аорты, общий артериальный ствол и тетрада Фалло).
- Иммунологические нарушения встречаются в 77% случаев (чаще поражается Т-клеточное звено, что проявляется предрасположенностью к грибковым заболеваниям, пневмоцистной инфекции, некоторым бактериальным и вирусным инфекциям).
- Гипокальциемия/гипопаратиреоз может проявляться судорожным синдромом при выраженном дефиците кальция в младенческом возрасте, существует риск развития нефрокальциноза. В детском возрасте и у 20% взрослых развивается гипотиреоз, в детском возрасте иногда встречается гипертиреоз.
- Поражение носоглоточного аппарата выявлено примерно в 70% случаев и проявляется в виде велофарингеальной недостаточности, расщеплении нёба, губы, раздвоении уздечки нёба, гнусавым оттенком голоса, также описано снижение обоняния, кондуктивная и/или сенсоневральная тугоухость. Вследствие велофарингеальной недостаточности у 36% пациентов отмечается затруднение глотания.
- Характерные черты лица (удлиненное лицо, микрогнатия или ретрогнатия, широкая переносица, мелкие зубы, асимметрия лица при плаче, опущенные вниз уголки рта, глазной гипертелоризм, низко посаженные и деформированные ушные раковины, бульбообразный кончик носа).
- Предрасположенность к аутоиммунным заболеваниям вследствие нарушения выработки Т-клеток (ювенильный ревматоидный артрит, аутоиммунная гемолитическая анемия, воспалительные заболевания кишечника, болезнь Грейвса, аутоиммунный увеит, бронхиальная астма).
- Задержка физического, речевого и психомоторного развития наблюдается у 70-90% пациентов.
- Поведенческие и психические проблемы (в детском возрасте обычно отмечаются гиперактивность, тревожность, аутические и аффективные расстройства; в подростковом и взрослом возрасте у 10-30% отмечаются биполярные расстройства, шизофрения, шизоаффективные расстройства.
- Повышен риск раннего старта (до 50 лет) болезни Паркинсона.
Синдром Бараката (Barakat syndrome, HDR-синдром (hypoparathyroidism, deafness and renal dysplasia syndrome) – редкое наследственное многокомпонентное заболевание, вызванное мутацией в гене GATA3, локализованном в коротком плече 10-й хромосомы (10р15) и кодирующем белок, отвечающий за развитие ОЩЖ, внутреннего уха, тимуса, почек, ЦНС. Впервые синдром описан американским доктором A. Дж. Баракатом в 1977 г. Описаны случаи как аутосомно-доминантного, так и аутосомно-рецессивного наследования синдрома. Заболевание характеризуется наличием у пациентов гипопаратиреоза (H - hypoprathyroidism, 83,2%), нейросенсорной тугоухости (D - deafness, 96,9%) и хронической болезни почек (R - renal disease, 65,1%). Поражение почек может иметь гетерогенный характер: различные пороки развития почек, нефротический синдром, гематурия, протеинурия, почечный тубулярный ацидоз. Другими проявлениями могут быть: нарушение работы эндокринной и репродуктивной систем (гипергонадотропный гипогонадизм, синдром поликистозных яичников, гипопитуитаризм), патология нервной системы (синдром Фара, когнитивные нарушения, аутизм, ишемические инсульты), врожденные пороки сердца и пороки развития лицевого черепа, пигментный ретинит. Пациенты чаще невысокого роста, что может быть связано с дефицитом соматотропного гормона. Уровень ПТГ колеблется от нижней границы нормы до неопределяемого.
Синдром Саньяда-Сакати (синдром Санджада-Сакати, Sanjad-Sakati syndrome, Middle East syndrome, синдром HRD (hypoparathyroidism retardation dysmorphism)) - редкое аутосомно-рецессивное генетическое заболевание, описанное в Саудовской Аравии в 1988 году и встречающееся у детей ближневосточного происхождения. Синдром Саньяда-Сакати часто рассматривается как вариант синдрома Кенни-Каффи 1 типа. Синдром Кенни-Каффи 1 типа и синдром Саньяда-Сакати являются следствием мутации в гене тубулин-специфичного шаперона Е (TBCE, tubulin-specific chaperone E) в локусе, который кодирует белок, отвечающий за связывание тубулина. Заболевание характеризуется врожденным гипопаратиреозом, выраженной задержкой роста, отставанием в умственном развитии, микроцефалией и лицевым дисморфизмом (длинное лицо, выступающий лоб, двустороннее опущение век, преаурикулярный отросток с правой стороны, клювовидный нос, вдавленная переносица, неправильный прикус, микрогнатия и микроцефалия).
Синдром Кенни-Каффи 1 типа (Kenny-Caffey syndrome 1) так же, как и синдром Саньяда-Сакати характеризуется врожденным гипопаратиреозом, тяжелой задержкой физического и психомоторного развития, микроцефалией, лицевым дисморфизмом (восходящий наклон глаз, тонкие губы, недоразвитые челюсти (микрогнатия), плоская переносица (придающая носу вид «клювовидного носа»), выступающий лоб и низко расположенные уши), но также наблюдаются остеосклероз и иммунодефицит. Аутосомно-рецессивное наследование синдрома.
Синдром Кенни-Каффи 2 типа (Kenny-Caffey syndrome 2) – редкое генетическое заболевание, поражающее преимущественно скелет, глаза, также встречаются различные патологии органов головы и шеи. Причиной являются мутации в гене FAM111А (family with sequence similarity 111 member A) на хромосоме 11q12, кодирующий белок-протеазу FAM111A, принимающей участие в репликации ДНК. Наследование синдрома аутосомно-доминантное. Частота встречаемости не известна, в литературе зарегистрировано менее 60 подтвержденных случаев. Одним из частых компонентов заболевания (диагностируется у 30-79% пациентов) является гипопаратиреоз. Характерными составляющими синдрома являются выраженная пропорциональная низкорослость, утолщение коры и стеноз мозгового вещества трубчатых костей, задержка закрытия большого родничка, аномалии глаз, гипопаратиреоз, нормальный интеллект.
Митохондриальные заболевания
Гипопаратиреоз является компонентом многих митохондриальных заболеваний, возникающих вследствие делеций в митохондриальной ДНК. Несмотря на то, что при данных заболеваниях гипопаратиреоз, как правило, не выступает основным симптомом, в ряде случаев в дебюте развиваются именно гипокальциемические судороги, что служит поводом для дальнейшего обследования.
Синдром Кернса-Сейра (Kearns-Sayre syndrome, KSS) – митохондриальная цитопатия, характеризующаяся энцефалопатией, офтальмоплегией (слабость глазодвигательных мышц с опущением век), пигментной ретинопатией и кардиомиопатией. Распространенность синдрома составляет 1-3 случая на 100 000 человек. В большинстве случаев синдром Кернса-Сейра приобретенный, т.е. мутации не передаются от родителей, а возникают после зачатия. Начало заболевания до 20 лет.
Синдром MELAS (mitochondrial encephalopathy, lactic acidosis and stroke-like episodes) – прогрессирующее нейродегенеративное заболевание, характеризующееся митохондриальной миопатией, энцефалопатией с эпилептическими приступами и/или деменцией, лактат-ацидозом, инсультоподобными эпизодами, непереносимостью физических нагрузок, мигренеподобной головной болью с тошнотой и рвотой. Впервые это заболевание было описано в 1984 году. Возникает вследствие мутации в митохондриальной транспортной рибонуклеиновой кислоте (тРНК). Чаще всего (80-90% случаев) обнаруживается мутация A3243G в гене MTTL1 тРНК аминокислоты лейцина. Синдром MELAS наследуется по материнской линии с началом заболевания до 40 лет. По разным данным, патология встречается с частотой от 1:15 000 до 1:20 000 человек. Заболевание чаще манифестирует в детстве (средний возраст 6-10 лет), после относительно благополучного периода раннего развития.
Дефицит митохондриального трифункционального белка (Mitochondrial trifunctional protein deficiency, MTPD syndrome, MTPDS) – аутосомно-рецессивное нарушение окисления жирных кислот, которое не позволяет организму преобразовывать определенные жиры в энергию, особенно в периоды без пищи. Дефицит митохондриального трифункционального белка может проявиться в младенчестве в виде низкого уровня сахара в крови, скелетной миопатии, дисфункции печени и задержки в развитии. Младенцы с этим заболеванием подвержены риску возникновения кардиомиопатии, затрудненного дыхания и пигментной ретинопатии. Признаки и симптомы MTPDS также могут включать гипотонию, мышечные боли, разрушение мышечной ткани и потерю чувствительности в конечностях, называемую периферической нейропатией. У некоторых пациентов с дефицитом MTP наблюдается прогрессирующее течение заболевания, связанное с миопатией и рецидивирующим рабдомиолизом.
Сниженный синтез или секреция паратгормона
Аутосомно-доминантная гипокальциемия (АДГ). При АДГ развивается умеренная гипокальциемия, но при стрессе может быть тяжелая гипокальциемия.
- АДГ 1 типа характеризуется активирующей мутацией в гене кальций-чувствительного рецептора CASR, который становится сверхчувствительным к сывороточным концентрациям кальция, а синтез и секреция ПТГ подавляются, несмотря на нормокальциемию. Возникает функциональный гипопаратиреоз. Почечная реабсорбция кальция при АДГ 1 типа снижена вследствие мутации CaSR в почках; у таких пациентов развивается гиперкальциурия.
- АДГ 2 типа возникает при наличии активирующей мутации в гене GNA11 (guanine nucleotide-binding protein alpha 11), который кодирует альфа-субъединицу G-белка G11, что приводит к избыточному подавлению секреции ПТГ даже при гипокальциемии. Почечная реабсорбция кальция при АДГ 2 типа не страдает.
- Синдром Барттера с гипокальциемией – один из редких вариантов манифестации АДГ, характеризуется гипокальциемией, гипомагниемией и гипопаратиреозом, а также признаками дисфункции петли Генле (полиурией, гипокалиемическим алкалозом, повышением концентрации ренина и альдостерона в плазме, низким артериальным давлением и резистентностью сосудов к ангиотензину II). Причиной развития синдрома является мутация в локусе 3q21.1, отвечающем за CaSR.
- Мутации в гене паратгормона. В литературе описано несколько редких мутаций в гене PTH, нарушающих его синтез и секрецию ПТГ. Например, аутосомно-доминантная мутация в сигнальной последовательности препроПТГ нарушает нормальный процессинг молекулы и, следовательно, образование ПТГ. Также описаны семьи с изолированным гипопаратиреозом, обусловленным аутосомно-рецессивной мутацией в гене PTH.
- Антитела к гену CaSR. При позднем развитии гипопаратиреоза возможной причиной могут являться циркулирующие активирующие антитела к гену кальций-чувствительного рецептора CaSR. Эти антитела не приводят к необратимой деструкции ОЩЖ, поэтому существует вероятность спонтанной ремиссии. Описаны как изолированные формы гипопаратиреоза, вызванного антителами к CaSR, так и случаи, ассоциированные c другими аутоиммунными заболеваниями (тиреоидитом Хашимото, болезнью Аддисона).
Синдромы резистентности к паратгормону
Псевдогипопаратиреоз (псевдоГПТ) - редкое наследственное заболевание, характеризующееся резистентностью органов-мишеней к ПТГ, гипокальциемией, повышением функции околощитовидных желези уровня ПТГ в крови, различными дефектами роста (низкорослость) и развития скелета (укорочение пястных и плюсневых костей), а также другими особенностями фенотипа. Наблюдается гипокальциемия, гиперфосфатемия на фоне повышенного уровня ПТГ. Нормализация уровня кальция при псевдоГПТ обычно приводит к снижению уровня ПТГ, но не устраняет резистентность тканей-мишеней к ПТГ. Распространенность псевдоГПТ составляет 1:100 000 – 1:295 000. Соотношение женщин и мужчин составляет 2:1, что указывает на возможность сцепленного с Х-хромосомой доминантного наследования. Патогенетически выделяют псевдогиперпаратиреоз 1 (1а, 1b, 1с) и 2 типов.
ПсевдоГПТ 1 типа является следствием мутации в гене GNAS1, кодирующем альфа-субъединицу стимулирующего G-белка, ассоциированного с рецептором ПТГ. Это приводит к неспособности G-белка активировать аденилатциклазу после связывания ПТГ с его рецептором и к невозможности трансдукции сигнала в органах-мишенях. Клинические проявления заболевания зависят от локализации мутации в материнском или отцовском аллеле, в связи с чем выделяют несколько подвидов псевдоГПТ 1 типа.
- ПсевдоГПТ тип 1а – семейное заболевание с аутосомно-доминантным типом наследования по материнской линии, изредка случаются спорадические случаи. ПсевдоГПТ 1а типа обусловлен мутацией гена GNAS1 на 20-й хромосоме, кодирующего α-субъединицу регуляторного белка, связывающего гуаниновые нуклеотиды (Gsα). Этот регуляторный белок служит посредником между рецепторами гормонов и аденилатциклазой. Мутантные Gsα не активируют аденилатциклазу. При псевдоГПТ активность Gsα в 2 раза ниже, чем в норме и реакция клеток-мишеней на ПТГ ослаблена. Чаще псевдоГПТ 1а типа проявляется наследственной остеодистрофией Олбрайта (F.Albright): низким ростом, ожирением, короткой шеей, округлым лицом, подкожными кальцинатами, укороченными четвертыми и пятыми метакарпальными и метатарзальными костями и отставанием в развитии. Помимо резистентности к ПТГ у больных может быть резистентность к другим гормонам, действие которых реализуется через G-белки: гонадотропин-рилизинг гормону, фолликулостимулирующегому гормону (ФСГ), лютеинизирующему гормону (ЛГ) и тиреотропному гормону (ТТГ). В биохимическом анализе крови наблюдается гипокальциемия с гиперфосфатемией в результате резистентности почек к действию ПТГ, и вторичный гиперпаратиреоз. Может развиваться фиброзный остеит, поскольку резистентность к ПТГ не затрагивает скелет.
- ПсевдоГПТ типа 1b – редкое аутосомно-доминантное заболевание, передающееся по материнской линии, в результате мутации регуляторных элементов гена GNAS1. Предполагают, что этот вариант заболевания обусловлен дефектом рецептора ПТГ. Активность Gsα нормальная. Клинических признаков синдрома Олбрайта нет. Биохимические признаки резистентности такие же, как при псевдоГПТ 1а типа (гипокальциемия, гиперфосфатемия и вторичный гиперпаратиреоз).
- ПсевдоГПТ типа 1с – редкое аутосомно-доминантное заболевание вследствием мутации, нарушающей связывание G-белка с рецептором ПТГ. Таким образом, G-белок способен активировать аденилатциклазу, но этот процесс не связан с активацией рецептора ПТГ. Активность Gsα нормальная. Наблюдаются признаки наследственной остеодистрофии Олбрайта (симптомы гипокальциемии; низкорослость; брахидактилия; лунообразное лицо; ожирение; крыловидные складки на шее; множественные подкожные кальцификаты; нередко умственная отсталость) и резистентность ко многим другим гормонам, аналогично ситуацией при псевдоГПТ 1а типа. Биохимические признаки резистентности такие же, как при псевдо ГПТ 1а типа.
ПсевдоГПТ типа 2 - редкое аутосомно-доминантное заболевание с наследственным нарушением метаболизма витамина D. Характерны гипокальциемия и гиперфосфатемия, но нет признаков наследственной остеодистрофии Олбрайта. Обнаруживается у пациентов с остеомаляцией, обусловленной дефицитом витамина D. Характерно повышение концентрации циклического аденозинмонофосфата (цАМФ) в моче при введении ПТГ без повышения концентрации фосфата в моче.
Псевдопсевдогиперпаратиреоз (псевдопсевдоГПТ) характеризуется снижением активности GSα, отсутствием резистентности к ПТГ, нормальными уровнями кальция и фосфора к крови, повышением уровня нефрогенного цАМФ в моче после введения ПТГ, наследственной остеодистрофией Олбрайта. При псевдопсевдоГПТ генетический дефект GSα не приводит к развитию полной картины резистентности к ПТГ, характерной для псевдоГПТ типа Iа. ПсевдопсевдоГПТ проявляется фенокопией псевдоГПТ 1а типа (симптомы гипокальциемии; низкорослость; брахидактилия; лунообразное лицо; ожирение; крыловидные складки на шее; множественные подкожные кальцификаты; нередко умственная отсталость) без его биохимических маркеров (сывороточные уровни кальция, фосфора и ПТГ остаются в пределах нормы).
Прогрессирующая костная гетероплазия вызывается унаследованной по отцовской линии инактивацией GNAS1, дебютирует в раннем детстве и характеризуется эктопическим костеобразованием в дерме, мышцах и соединительной ткани. В клинической картине могут наблюдаться признаки, характерные для наследственной остеодистрофии Олбрайта, не сопровождающиеся изменением концентрация кальция или ПТГ в сыворотке крови.
Гипопаратиреоз и гипомагниемия
Дефицит магния в сыворотке крови приводит к патологическому снижению уровня ПТГ и развитию резистентности костной ткани и почек к его эффектам. Гипомагниемия наблюдается вследствие: недостаточного поступления с пищей; снижения абсорбции в кишечнике (мальабсорбция, синдром короткой кишки, тяжелая и продолжительная рвота, диарея, стеаторея); семейной гипомагниемии с вторичной гипокальциемией (мутация TRPM6); аутосомно-доминантной гипокальциемии (активирующая мутация в гене CASR); семейной гипомагниемии с гиперкальциурией и нефрокальцинозом (FHHNC); приема лекарственных препаратов (тиазиды, фуросемид, ингибиторы протонного насоса, антибактериальные препараты системного действия, противогрибковые препараты системного действия, противоопухолевые препараты, иммунодепрессанты, моноклональные антитела и конъюгаты антител ). Гипермагниемия также может приводить к нарушению функции ОЩЖ путем подавления секреции ПТГ с развитием гипокальциемии. Гипермагниемия может развиться при следующих состояниях: хронической болезни почек (ХБП); семейной гипокальциурической гиперкальциемии; избыточном потреблении (парентеральное введение препаратов магния и др.); токолитической терапии при эклампсии.
Клиническая картина гипопаратиреоза
Основные клинические проявления гипопаратиреоза обусловлены гипокальциемией, связаны с изменениями мембранного потенциала клеток, обусловливающими нейромышечную возбудимость, и дисфункцией нервной и нервно-мышечной систем. Недостаток кальция приводит к частичной деполяризации потенциала покоя на мембране нейронов, увеличивая вероятность запуска потенциала действия и, как следствие повышается нервно-мышечная возбудимость и общая вегетативная реактивность. Степень выраженности симптомов зависит от уровня кальция в сыворотке крови, а также от скорости прогрессирования гипокальциемии. Усиление чувствительности сенсорного (чувствительного) нейрона проявляется в виде парестезий в конечностях и в околоротовой области; моторного (двигательного) нейрона – мышечными спазмами, вплоть до тетании от классического карпопедального спазма до жизнеугрожающего ларингоспазма; тяжелая гипокальциемия ассоциирована с локальными или генерализованными судорогами тонико-клонического типа.
Гипопаратиреоз может быть латентным, который клинически не проявляется и выявляется при лабораторном исследовании, или манифестным, протекающий с клиническими проявлениями. Манифестный гипопаратиреоз по течению может быть острым или хроническим.
Клинические проявления хронического гипопаратиреоза. Хронический гипопаратиреоз часто протекает бессимптомно. В клинической картине манифестного гипопаратиреоза выделяют 4 основные группы синдромов:
- Тетанический, судорожный синдром (повышенная нервно мышечная возбудимость).
- Синдром изменений чувствительной сферы и вегетативных функций.
- Синдром поражения центральной нервной системы, мозговых нарушений.
- Изменения кожи и трофические нарушения.
Тетанический, судорожный синдром (тетания, тетанус). Судорогам предшествуют предвестники - парестезии, жжение, покалывание, напряжение, скованность в мышцах рук и ног. Затем развиваются тонические и клонические судороги отдельных групп мышц. Для судорог характерен избирательный характер, они распространяются на отдельные группы мышц, симметрично с той и другой стороны. Чаще всего вовлекаются преимущественно симметричные группы сгибательных мышц верхних конечностей, потом судороги распространяются на мышцы нижних конечностей. При судорогах верхних конечностей плечо приведено к туловищу, предплечье согнуто в локтевом суставе, кисть согнута в локтевом, лучезапястном и пястно-запястном суставах, пальцы сжаты и склонены к ладони («рука акушера»). При судорогах мышц нижних конечностей стопа изогнута внутрь, пальцы в положении подошвенного сгибания, большой палец покрыт остальными и подошва вдавлена в виде желоба, ноги тесно прижаты одна к другой в вытянутом положении. Нередко вовлекаются мышцы лица, реже - диафрагмы, туловища, внутренних органов, гортани.
Судороги мышц лица придают ему характерное выражение: углы рта опускаются, рот приобретает форму «рыбьего» («сардоническая улыбка»), наблюдается спазм жевательной мускулатуры (тризм, жевательные мышцы очень напряжены), брови сдвинуты, веки полуопущены, наблюдаются судороги век. Судороги мышц туловища вызывают судорожное разгибание туловища кзади (опистотонус). Судороги межреберных мышц и диафрагмы приводят к затруднению дыхания. Судороги мышц гортани (ларингоспазм) - наиболее опасное проявление тетании; чаще встречаются у детей и проявляются инспираторной одышкой, шумным стенотическим дыханием, цианозом, на губах появляется пена; в тяжелых случаях больной теряет сознание и наступает смерть от асфиксии, если не сделаны срочная трахеотомия или интубация. Судороги мышц пищевода приводят к нарушению глотания. Судороги мышц привратника (пилороспазм) вызывают тошноту, рвоту, боли в эпигастрии. Спазм мускулатуры кишечника вызывает запоры, часто кишечную колику. Спазм мышц мочевого пузыря приводит к анурии. При спазме венечных артерий появляются резкие боли в области сердца, напоминающие стенокардию. Сознание при тетании сохранено, однако при особенно тяжелых приступах может нарушаться.
Приступы судорог могут быть от минут до нескольких часов; могут возникать как спонтанно, так и под влиянием различных раздражителей - механических, термических, электрических, болевых. Иногда судороги могут провоцироваться мышечным напряжением при физической нагрузке, нервным потрясением, горячей ванной, выпрямлением конечностей. Если преобладает тонус симпатической нервной системы, приступ протекает с бледностью из-за спазма периферических артерий, тахикардией, повышением АД. Если преобладает тонус парасимпатической нервной системы, типичны рвота, поносы, полиурия, брадикардия, гипотония. Как правило, судорожные явления и развернутая картина гипопаратиреоидного криза возникают при снижении содержания кальция в крови до 1,9-2,0 ммоль/л. Тетания может быть как явной, проявляющейся спонтанными симптомами, так и латентной, которая выявляется только с помощью провокационных манипуляций. Латентная тетания обычно встречается при меньшем снижении концентрации кальция в сыворотке; 7-8 мг/дл (1,75-2,20 ммоль/л).
Латентную тетанию легко обнаружить у больного, вызывая у него симптомы Хвостека, Труссо, Вейса и др.
Симптом Хвостека – непроизвольное сокращение мышц лица при легком постукивании в области лицевого нерва ~2 см кпереди от мочки уха, чуть ниже скуловой дуги. Различают симптом Хвостека I степени, когда сокращаются все мышцы лица на стороне постукивания; II степени - сокращаются мышцы в области крыльев носа и угла рта; III степени - только в области угла рта.
Симптом Труссо – судороги в области кисти («рука акушера», «пишущая рука»), возникающие после компрессии плеча в течение 3 минут манжетой тонометра, накаченного на 20 мм рт. ст. выше систолического артериального давления.
Симптом Вейса - сокращение мышцы век и лобной мышцы при поколачивании у наружного края глазницы.
Симптом Гофмана - появление парестезии при надавливании в участках разветвления нервов, в частности, в месте выхода 1 ветви тройничного нерва (у внутреннего края брови).
Симптом Шлезингера - судороги в разгибательных мышцах бедра и стопы при быстром пассивном сгибании ноги в тазобедренном суставе при выпрямленном коленном суставе.
Симптом Эрба - повышенная электровозбудимость нервов конечностей при раздражении слабым гальваническим током (<0,5 мА), что выражается в судорогах.
Синдром изменений чувствительной сферы и вегетативных функций. Вне приступов тетании вегетативные нарушения у больных проявляются ощущением похолодания или жара, потливостью, головокружением, обмороками, нарушением зрительной аккомодации, диплопией, мигренью, звоном в ушах, иногда болями, перебоями в области сердца, уменьшением сумеречного зрения. Изменения чувствительной сферы характеризуются повышенной чувствительностью к шуму, резким звукам, громкой музыке, изменением восприятия температуры окружающей среды (когда всем тепло, больному холодно и наоборот), уменьшением чувствительности к кислому, повышением чувствительности к горькому, сладкому.
Синдром поражения центральной нервной системы, мозговых нарушений. При длительной гипокальциемии развиваются изменения психики, неврозы, эмоциональные нарушения (депрессия, тоска), бессонница. В тяжелых приступах тетании может быть отек мозга со стволовыми и экстрапирамидными симптомами. Мозговые нарушения при гипотиреозе могут проявляться эпилептиформными приступами. В отличие от классической эпилепсии наблюдается быстрая благоприятная динамика ЭЭГ при достижении стойкой нормокальциемии, купирующий эффект внутривенного введения препаратов кальция, а также отсутствие потери сознания. Наиболее тяжелые неврологические изменения наблюдаются у больных с внутричерепной кальцификацией, которая выявляется при гипопаратиреозе в 52-74% случаев, чаще в области базальных ганглиев, а также над турецким седлом, иногда - в области мозжечка. Проявления, связанные с внутричерепной кальцификацией, полиморфны и зависят от локализации кальцификатов и степени повышения внутричерепного давления: чаще наблюдаются явления эпилептиформного типа и паркинсонизм. Поражение центральной нервной системы при гипопаратиреозе может протекать в виде синдрома Фара, который проявляется: • обызвествлением в области базальных ганглиев мозга; • эпилепсией; • деменцией; • головными болями; • экстрапирамидными знаками.
Изменения кожи и трофические нарушения. Характерны сухость и шелушение кожи (52%), экзема, эксфолиативный дерматит, возможны пигментации, очаги депигментации кожи (витилиго), очень часто развивается кандидомикоз. Возможно появление на коже пузырей с жидким содержимым. Поражаются также придатки кожи: наблюдаются нарушения роста волос, раннее поседение и поредение волос (52-62%), иногда наблюдается их выпадение вплоть до полной алопеции (10%), ломкость, бледность, тусклость ногтей (29%), частое их грибковое поражение. Изменения кожи могут быть моносимптомом гипопаратиреоза. К трофическим нарушениям относятся также изменения зубочелюстной системы: у детей - нарушения формирования зубов, дефекты эмали; у больных всех возрастных групп - кариес, дефекты эмали зубов. Характерное проявление гипопаратиреоза у детей - задержка роста. Одно из нарушений трофики у больных гипопаратиреозом - гипокальциемическая катаракта (27-55%). Возможна врожденная катаракта (при тетании матери или врожденном гипопаратиреозе).
Сердечно-сосудистая система. Гипокальциемия способствует нарушению сердечного ритма. У пациентов с хронической гипокальциемией на ЭКГ может выявляться удлинение интервала QT, наряду с изменениями S и Т зубцов. Большинство из этих симптомов разрешаются на фоне компенсации заболевания и достижения стойкой нормокальциемии. Формирующаяся при длительном тяжелом течении гипопаратиреоза гипокальциемическая дилатационная кардиомиопатия, также, как правило, обратима на фоне компенсации заболевания. Пациенты с хроническим гипопаратиреозом имеют повышенные риски фибрилляции предсердий, тахиаритмий, инфаркта миокарда, ишемической болезни сердца, сердечной недостаточности, инсульта, цереброваскулярных заболеваний и заболеваний периферических сосудов.
Почки. Традиционное лечение препаратами кальция и витамина D и его аналогов способствует повышенной экскреции кальция с мочой из-за отсутствия ПТГ-опосредованной реабсорбции кальция в дистальном канальце нефрона. Гиперкальциурия является фактором риска нефрокальциноза и нефролитиаза, ассоциированных с развитием хронической болезни почек и хронической почечной недостаточности вплоть до терминальной стадии. Частота развития нефролитиаза/нефрокальциноза у пациентов, получающих лечение препаратами кальция и витамина D, варьирует от 12% до 57%, при этом риски манифестации в 5 раз выше общепопуляционных показателей. Кроме того, имеются данные о значимом повышении риска почечной недостаточности.
Острая гипокальциемия (гипокальциемический криз)
Острая гипокальциемия (гипокальциемический криз) – это жизнеугрожающее состояние, которое требует неотложных лечебных мероприятий. Клинически она проявляется приступами тетании, которым могут предшествовать парестезии лица, кистей и стоп, чувство страха, беспокойство, фибриллярные подергивания отдельных мышц. Чаще всего судороги возникают в мышцах верхних конечностей, реже – в нижних. Вследствие спазма мускулатуры лица возникает сардоническая улыбка, губы приобретают форму «рыбьего рта». При спазме жевательных мышц возникает судорожное сжатие челюстей (тризм). Судороги в мышцах верхних конечностей приводят к характерному положению руки, называемому «рукой акушера»: пальцы сжаты и слегка приведены к ладони, I палец сведен, кисть согнута в лучезапястном суставе. При спазме мускулатуры нижних конечностей бедра и голени вытянуты, стопы поворачиваются внутрь, туловище выгибается кзади (опистотонус). Вследствие судорожных сокращений межреберных мышц, мышц живота и диафрагмы резко нарушается дыхание. Изменения органов и систем при тетании зависят от преобладания тонуса симпатической или парасимпатической системы. Активация симпатической нервной системы приводит к тахикардии, повышению артериального давления, побледнению кожных покровов вследствие спазма периферических сосудов, а парасимпатической - к брадикардии и гипотензии, рвоте, диарее, полиурии. Спазмы гладкой мускулатуры внутренних органов и сосудов могут имитировать различные заболевания, включая патологию сердечно-сосудистой системы (приступы стенокардии, эндартериита, мигрени и т.д.), органов дыхания (например, бронхиальная астма), желудочно-кишечного тракта (холецистит, панкреатит, аппендицит, язва желудка), мочеполовой системы (цистит, нефрит и др.).
Лабораторная диагностика гипопаратиреоза
- Исследование уровня общего кальция и ионизированного кальция в крови. Диагноз гипопаратиреоза устанавливается на основании выявления гипокальциемии в сочетании со снижением уровня ПТГ (или выявлением неадекватно низкого уровня ПТГ). Диагноз гипокальциемии устанавливают при концентрации общего кальция в сыворотке <8,8 мг/дл (<2,2 ммоль/л) при норме 2,25-2,75 ммоль/л; концентрации ионизированного кальция в сыворотке <4,7 мг/дл (<1,17 ммоль/л) является низкой при норме 1,03-1,24 ммоль/л.
- Исследование уровня альбумина в крови и расчет альбумин-скорректированного кальция. При оценке содержания общего кальция крови для исключения ложноположительных и ложноотрицательных результатов следует вносить поправку в зависимости от уровня альбумина с расчетом альбумин-скорректированного кальция. При содержании альбумина сыворотки <40 г/л к определяемому уровню кальция добавляют 0,1 ммоль/л на каждые недостающие 6 г/л альбумина. При содержании альбумина >40 г/л вычитают 0,1 ммоль/л на каждые избыточные 6 г/л альбумина.
- Исследование уровня паратгормона в крови. В норме содержание паратгормона в крови – 0,15-0,6 мг/мл.
- Исследование уровня фосфора в крови. При гипопаратиреозе наблюдается повышение уровня фосфора в крови при норме 0,64-1,29 ммоль/л.
- Исследование уровня магния в крови. При гипомагниемии снижается секреция ПТГ и повышается резистентность к ПТГ костей и почек.
- Исследование суточной экскреции кальция с мочой. При гипокальциемии наблюдается гипокальциурия при норме 200-400 мг кальция в сутки.
- Исследование суточной экскреции фосфора с мочой. При гипопаратиреозе экскреция фосфора с мочой снижается (гипофосфатурия).
- Исследование уровня 25(ОН)D витамина D в крови. Тяжелый дефицит витамина D может быть ассоциирован с гипокальциемией. Определение уровня активного метаболита 1,25(ОН)2D не показано, так как данный метаболит имеет короткий период полувыведения и не отражает статуса витамина D в организме.
- Исследование уровня креатинина в крови с расчетом СКФ. Гипокальциемия часто наблюдается при снижении скорости клубочковой фильтрации (СКФ).
Для оценки адекватности терапии при гипопаратиреозе рекомендован динамический мониторинг уровней общего кальция в крови, альбумина в крови (с расчетом альбумин-скорректированного кальция), ионизированного кальция в крови, фосфора в крови, креатинина в крови с расчетом СКФ, а также уровня кальция в суточной моче. В случае компенсации заболевания мониторинг проводится с частотой 1 раз в 3-6 месяцев. При отсутствии компенсации гипопаратиреоза рекомендована более частая оценка показателей фосфорно-кальциевого обмена, до нескольких раз в неделю. Мониторинг уровня кальция в суточной моче проводится 1 раз в 6-12 месяцев. В случае выявления гиперкальциурии и/или назначении тиазидов или тиазидоподобных диуретиков определение уровня кальция в суточной моче рекомендовано выполнить через 1,5-2 месяца для оценки адекватности проводимого лечения.
Инструментальные исследования при гипопаратиреозе
ЭКГ позволяет выявить нарушения сердечного ритма и удлинение интервалов QT и ST. Пациентам с гипопаратиреозом рекомендуется 1 раз в год УЗИ почек и/или КТ почек и надпочечников в связи с повышенным риском нефролитиаза/нефрокальциноза. При хроническом гипопаратиреозе и терапии препаратами витамина D и его аналогов, препаратами кальция риск развития нефролитиаза/нефрокальциноза значимо повышается. При длительности заболевания более 10 лет в связи с повышенным риском катаракты рекомендовано проведение комплексного офтальмологического обследования. При наличии неврологических симптомов при гипопаратиреозе рекомендуется проведение КТ головного мозга и консультация врача-невролога. Клинические проявления кальцификации различных отделов центральной нервной системы у пациентов с длительным анамнезом гипопаратиреоза неспецифичны. К наиболее распространенным относятся двигательные нарушения: ригидность мышц, паркинсонизм, гиперкинезы (хорея, тремор, дистония, атетоз, орофациальная дискинезия); когнитивные расстройства; мозжечковые симптомы и нарушения речи. В ряде случаев отмечаются эпилептические приступы, деменция. При гипопаратиреозе неуточненной этиологии рекомендуется генетическое тестирование и консультирование для исключения наследственного характера патологии. При изолированном гипопаратиреозе неясной этиологии, возникшим после первого года жизни, рекомендуется исследование гена AIRE для исключения АПС 1-го типа (исходно оценка мутации R257X, в случае отрицательного результата – секвенирование гена). Диагноз АПС 1 типа может быть установлен клинически при наличии у пациента как минимум двух из трех «основных» компонентов заболевания («классическая диада или триада»), а также при наличии одного из них в случае присутствия заболевания у родственника первого порядка. С целью верификации синдрома также может быть использовано определение антител к интерферону 1 типа, особенно к ИНФ-ω и к ИНФ-α2, предложенных в качестве диагностических критериев заболевания. Определение антител к 21-гидроксилазе полезно для прогнозирования развития надпочечниковой недостаточности.
Лечение гипопаратиреоза
Терапия гипопаратиреоза и псевдогипопаратиреоза направлена на повышение содержания кальция и снижение концентрации неорганического фосфора в крови. Цели лечения: восстановление фосфорно-кальциевого баланса в организме, поддержание нормального уровня кальция и фосфора крови, улучшение качества жизни.
Немедикаментозное лечение: Рацион питания пациента должен включать продукты с высоким содержанием кальция (молочные продукты, зелень, кунжут и др.); В определённых случаях требуется ограничить продукты, содержащие фосфаты (мясо, субпродукты, яйца); Обогатить рацион продуктами, содержащими большие количества витамина Д (печень трески, рыбий жир, сливочное масло).
Медикаментозное лечение: Комбинированная терапия препаратами витамина D и его аналогов (альфакальцидол 1,0-4,0 мкг/сутки, кальцитриол 0,25-2,0 мкг/сутки) и нативной формы витамина D (колекальциферол 800-1000 МЕ). Препараты солей кальция (чаще всего карбонат кальция) - назначаются для восполнения дефицита иона кальция в организме (средняя суточная доза препаратов кальция составляет 1000-2000 мг и требует тщательного индивидуального подбора). Лечение гипокальциемии, вызванной дефицитом магния или сопровождающееся им, требует применения препаратов магния в сочетании с витамином D и солями кальция. При хроническим гипопаратиреозе с гиперкальциурией рекомендована терапия тиазидами и тиазидоподобными диуретиками - сульфонамидами (хлорталидон 1 раз в сутки внутрь в дозе 25-200 мг) для достижения целевого уровня экскреции кальция. При транзиторном послеоперационном гипопаратиреозе часто достаточно пероральных препаратов кальция (глюконата кальция или карбоната кальция).
Лечение острой гипокальциемии
При острой гипокальциемии вначале внутривенно медленно вводят 10 мл 10% раствора кальция глюконата в 50-100 мл 0,9% раствора натрия хлорида (или в 5% раствора декстрозы) в течение 10-20 минут. Затем внутривенно капельно вводят 100 мл 10% раствора кальция глюконата со скоростью 0,5-1,5 мг/кг/час в 1000 мл 0,9% раствора натрия хлорида (или 5% раствора декстрозы) со скоростью 50 мл/час (в среднем в течение 8-10 часов). Одновременно назначаются пероральные препараты кальция и препараты витамина D и его аналогов ((альфакальцидол в среднем 3-3,5 мкг/сут и/или кальцитриол 1,5-2 мкг/сут). При наличии выраженной гипомагниемии показана ее коррекция с использованием как пероральных препаратов (препараты комбинации различных препаратов магния 300-400 мг/сут), так и внутривенных форм – внутривенно струйно 2 грамма магния сульфата в течение 10-20 минут, внутривенно капельно 25% раствор магния сульфата 2-4 грамма в 150-200 мл физиологического раствора натрия хлорида.
Автор - Иогансен Юрий Александрович, врач ультразвуковой диагностики, кандидат медицинских наук.