1.0 Введение: Фиброз Миокарда как Ключевая Терапевтическая Мишень при Сердечной Недостаточности
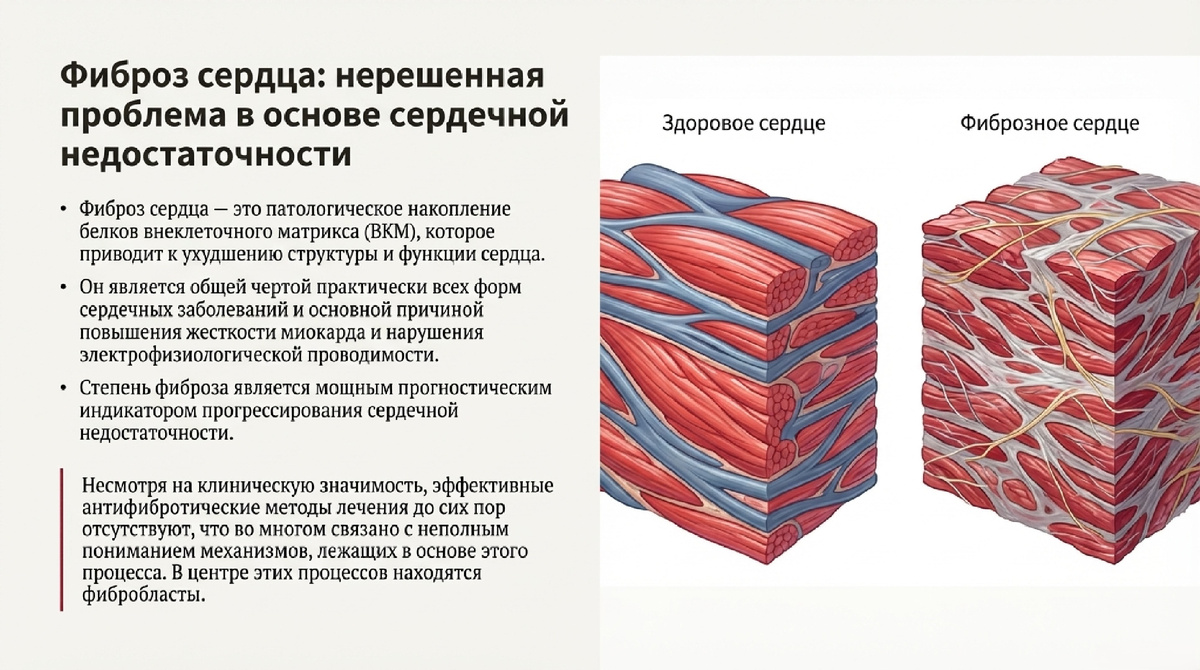
Фиброз миокарда представляет собой патологический процесс, характеризующийся избыточным накоплением белков внеклеточного матрикса (ECM), что приводит к структурной и функциональной деградации сердца. Стратегическая важность этого процесса обусловлена тем, что фиброз является общим признаком практически всех форм сердечных заболеваний и служит мощным прогностическим индикатором прогрессирования сердечной недостаточности. Накопление фиброзной ткани повышает жесткость миокарда и нарушает его электрофизиологическую проводимость, тем самым напрямую способствуя развитию сердечной недостаточности.
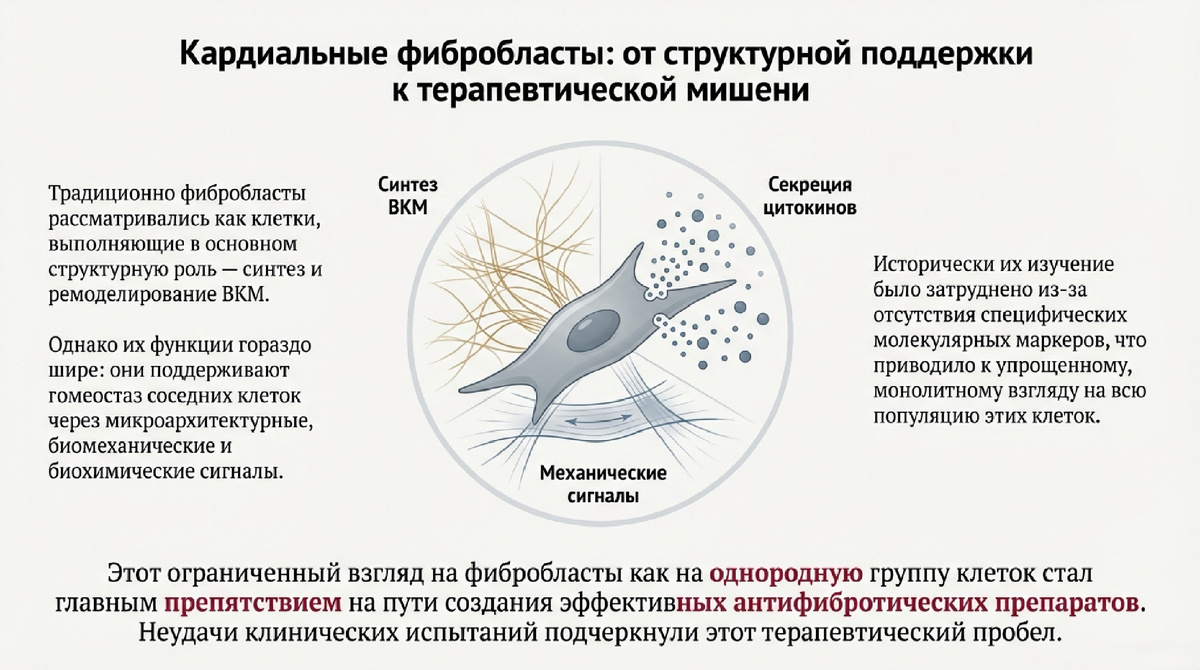
Центральную роль в этом процессе играют сердечные фибробласты — гетерогенная популяция стромальных клеток, которые являются основными клеточными медиаторами продукции ECM. В норме они поддерживают гомеостаз тканей, однако при повреждении или стрессе они активируются и запускают каскад фибротических реакций. Несмотря на клиническую значимость, на сегодняшний день не существует антифибротических терапевтических методов с доказанной долгосрочной эффективностью, что во многом связано с неполным пониманием молекулярных механизмов, лежащих в основе фиброгенеза.
Цель данного доклада — систематизировать и проанализировать современные и перспективные терапевтические стратегии, направленные на модуляцию активности фибробластов, основываясь на последних научных данных, полученных с помощью передовых технологий, таких как секвенирование РНК единичных клеток. Для разработки эффективной таргетной терапии необходимо более глубокое рассмотрение патогенеза фиброза, что позволит выявить ключевые уязвимости в механизмах его развития.
2.0 Патогенез Фиброза Миокарда и Ключевая Роль Фибробластов
Понимание различных клеточных состояний фибробластов и типов фиброзного ремоделирования является фундаментальной основой для разработки эффективных антифибротических методов лечения. Современные исследования позволили выделить четкие паттерны фиброза и охарактеризовать динамические переходы фибробластов из состояния покоя в активированные, профибротические фенотипы.
2.1 Типы Фиброзного Ремоделирования Сердца
В патологии сердца выделяют два основных типа фиброза, которые могут сосуществовать, но имеют различные причины и потенциал обратимости.
• Заместительный фиброз возникает как следствие гибели кардиомиоцитов, например, после инфаркта миокарда. В этом случае утраченные мышечные клетки замещаются плотной рубцовой тканью. Данный тип фиброза считается необратимым при отсутствии механизмов для эффективной регенерации миоцитов.
• Реактивный фиброз характеризуется диффузным накоплением ECM в интерстициальном или периваскулярном пространстве и развивается при ограниченной гибели миоцитов. Этот тип фиброза ассоциирован с такими состояниями, как артериальная гипертензия, сахарный диабет и сердечная недостаточность с сохраненной фракцией выброса (СНсФВ). Существует гипотеза, что реактивный фиброз потенциально обратим, особенно если коллагеновые волокна еще не подверглись избыточному перекрестному сшиванию.
2.2 Трансформация Фибробластов: от Покоя к Активации
При повреждении миокарда резидентные фибробласты переходят из состояния покоя в активированное состояние, а затем дифференцируются в миофибробласты. Этот процесс является центральным звеном фиброгенеза.
• Маркеры покоящихся фибробластов: Резидентные популяции фибробластов идентифицируются по экспрессии таких маркеров, как транскрипционный фактор 21 (Tcf21+) и рецептор тромбоцитарного фактора роста-α (PDGFR-α+).
• Маркеры активированных фибробластов и миофибробластов: Активированные клетки и миофибробласты экспрессируют матрицеллюлярный белок периостин (Postn). Классическим маркером, определяющим фенотип миофибробласта, остается альфа-актин гладких мышц (α-SMA, кодируемый геном ACTA2).
Ключевая функция миофибробластов заключается в интенсивной продукции и ремоделировании фиброзного ECM. Они синтезируют фибриллярные коллагены (в частности, Коллаген 1, Коллаген 3, Коллаген 5 и Коллаген 6) и ферменты, способствующие созреванию матрикса, такие как лизилоксидаза (Lox). Активность миофибробластов напрямую определяет масштаб и тяжесть фиброза.
2.3 Судьба Миофибробластов после Повреждения
После разрешения повреждающего стимула дальнейшая судьба миофибробластов может развиваться по нескольким сценариям, что бросает вызов ранее существовавшему мнению об их неизбежном апоптозе.
1. Реверсия в состояние покоя: Исследования на моделях гипертонии, вызванной ангиотензином II, показали, что часть миофибробластов может возвращаться в неактивное состояние. Однако такие клетки могут оставаться «подготовленными» к быстрой повторной активации при новом стрессе.
2. Де-дифференцировка в матрифиброциты: Этот процесс характерен для заживления после ишемического повреждения. Миофибробласты трансформируются в матрифиброциты, которые экспрессируют гены, связанные с формированием хрящевой и костной ткани (Sox9, Comp, Cilp, Runx1). Считается, что эта трансформация способствует стабилизации и созреванию рубца.
3. Персистенция: Миофибробласты могут сохраняться в миокарде в течение длительного времени даже после устранения первоначального стимула, поддерживая хронический фиброз. При этом важно отметить, что чем дольше поддерживается состояние миофибробласта, тем более стабильным оно становится, что определяет потенциальное терапевтическое окно для вмешательства.
Эта гетерогенность судеб миофибробластов — от персистенции до де-дифференцировки — подчеркивает необходимость разработки терапевтических стратегий, способных не просто блокировать их активацию, но и целенаправленно управлять их конечной судьбой, что является ключевой задачей для регенеративной кардиологии. Эти клеточные трансформации управляются сложной сетью молекулярных сигнальных каскадов, которые представляют собой перспективные мишени для терапевтического вмешательства.
3.0 Молекулярные Механизмы, Управляющие Активацией Фибробластов
Идентификация и глубокое понимание конкретных сигнальных путей, контролирующих состояние фибробластов, открывают прямые возможности для разработки таргетной фармакологической интервенции. Ниже рассмотрены ключевые молекулярные регуляторы, управляющие активацией, пролиферацией и механочувствительностью этих клеток.
3.1 Ключевые Профибротические Сигнальные Пути
• Сигнальный путь трансформирующего фактора роста-β (TGF-β): Этот путь является одним из центральных индукторов фиброза. Он разделяется на два основных каскада:
◦ Канонический путь (TGF-βR1/2-SMAD2/3): Играет ключевую роль в индукции и поддержании фенотипа миофибробластов.
◦ Неканонический путь (TGF-βR2-p38 MAPK): Также необходим для активации фибробластов. Его специфическое ингибирование в фибробластах значительно снижает фиброз.
• Эпигенетический регулятор BRD4: Является нижестоящим эффектором p38-сигналинга. Этот белок, связывающийся с ацетилированными гистонами, эпигенетически репрограммирует фибробласты, переводя их в профибротическое состояние. Ингибирование BRD4 с помощью малой молекулы JQ1 продемонстрировало способность снижать фиброз сердца.
• РНК-связывающий белок MBNL1: Был идентифицирован как ключевой посттранскрипционный «переключатель» между состояниями фибробласта и миофибробласта. Делеция гена MBNL1 в фибробластах приводила к снижению фиброзного рубцевания и способствовала формированию репаративного, а не фибротического фенотипа после ишемического повреждения.
3.2 Регуляция Пролиферации и Клеточного Цикла
Пролиферация фибробластов играет двойственную роль в фибротическом ремоделировании. Как правило, фибробласты пролиферируют в ответ на стресс еще до их полной активации в миофибробласты. Исследования показали, что активность клеточного цикла может подавлять немедленную активацию. Например, делеция гена-супрессора опухолей p53 в сердечных фибробластах приводит к усиленной пролиферации, что, как ни парадоксально, отсрочивает, но в конечном итоге усугубляет фиброз, поскольку фибробласты выходят из клеточного цикла и массово активируются позже.
3.3 Механочувствительная Регуляция и Положительная Обратная Связь
Фибробласты способны воспринимать и реагировать на механические свойства своего окружения, в частности, на жесткость внеклеточного матрикса. Этот процесс, известный как механотрансдукция, создает контур положительной обратной связи, который поддерживает и усиливает фиброз.
Ключевые механочувствительные пути включают:
• YAP/TAZ: Транскрипционные коактиваторы, играющие критическую роль в активации фибробластов в ответ на механические стимулы.
• Ионные каналы: Механочувствительные каналы, такие как Piezo1, TRPC6 и TRPV4, усиливают приток ионов кальция (Ca2+) в клетку, что способствует трансформации фибробластов в миофибробласты.
• Тирозинкиназа Src: Мета-анализ данных scRNA-seq позволил идентифицировать Src как обогащенный в фибробластах механосенсор, связанный с фокальными контактами.
Эти пути формируют «механический контур обратной связи»: чем жестче становится матрикс, тем сильнее активируются фибробласты, которые, в свою очередь, производят еще больше ECM, дополнительно увеличивая жесткость ткани. С клинической точки зрения, этот контур объясняет, почему фиброз может приобретать самоподдерживающийся и прогрессирующий характер даже после устранения первоначального повреждающего фактора, что представляет собой серьезную терапевтическую проблему. Фибробласты функционируют не в изоляции, а в тесной кооперации с другими типами клеток, прежде всего с клетками иммунной системы, что создает еще один уровень регуляции.
4.0 Взаимодействие Фибробластов с Иммунной Системой и Клетками Миокарда
Стратегическое значение межклеточных взаимодействий в сердце трудно переоценить. Фибробласты выступают в роли центральных узлов, координирующих как воспалительные, так и репаративные процессы. Они активно взаимодействуют с иммунными клетками, кардиомиоцитами и эндотелием, формируя сложную регуляторную сеть.
4.1 Фибробласты как Координаторы Воспалительного Ответа
В ответ на повреждение фибробласты функционируют как «иммунные стражи» сердца, модулируя активность врожденной и адаптивной иммунной системы.
• Взаимодействие с врожденной иммунной системой: Фибробласты экспрессируют Toll-подобные рецепторы, в частности TLR4, что позволяет им распознавать молекулярные паттерны, ассоциированные с повреждением (DAMPs), такие как белки S100A8/A9. Активация TLR4 запускает провоспалительные каскады, включая активацию инфламмасомы NLRP3, что ведет к секреции цитокинов IL-1β и IL-18 и дальнейшему усилению воспаления.
• Взаимодействие с адаптивной иммунной системой: Фибробласты способны представлять антигены через молекулы MHCII, напрямую взаимодействуя с CD4+ Т-клетками. Кроме того, они секретируют широкий спектр хемокинов (CCL20, CXCL9, CXCL10, CXCL12), которые привлекают в очаг повреждения различные субпопуляции Т-клеток, включая провоспалительные Th17-клетки и регуляторные Т-клетки. Следует отметить, что современные представления о центральной роли медиаторов, продуцируемых фибробластами, основаны преимущественно на исследованиях in vitro. Для подтверждения их причинной роли в формировании воспалительного фенотипа in vivo необходимы дальнейшие исследования с использованием моделей со специфической делецией соответствующих хемокинов в фибробластах.
Таким образом, в зависимости от контекста и микроокружения, фибробласты могут как усиливать воспалительную реакцию, так и способствовать ее разрешению, что подчеркивает их двойственную роль в иммунном ответе.
4.2 Кросс-ток с Кардиомиоцитами и Эндотелиальными Клетками
Фибробласты находятся в постоянном диалоге с другими ключевыми клетками миокарда, используя для этого биохимические, механические и электрические сигналы.
Взаимодействие с кардиомиоцитами:
• Биохимическое: Фибробласты и кардиомиоциты обмениваются растворимыми факторами (TGF-β, PDGF, Ang II) и внеклеточными везикулами (экзосомами), которые могут переносить регуляторные молекулы, такие как miR-208a (от миоцитов к фибробластам) и miR-21 (от фибробластов к миоцитам).
• Механическое/Электрическое: Прямой физический и электрический контакт между клетками возможен через щелевые контакты. Кроме того, передача механических сил осуществляется через ECM и фокальные контакты, что позволяет клеткам «чувствовать» сократительную активность друг друга.
Взаимодействие с эндотелиальными клетками:
1. Поддержка ангиогенеза: Фибробласты играют важную роль в росте и ремоделировании сосудов. Например, особый Wnt-фибробластный субстат находится в тесном контакте с эндотелиальными клетками в зоне инфаркта и способствует формированию новых сосудов.
2. Эндотелиально-мезенхимальный переход (EndMT): В определенных патологических условиях эндотелиальные клетки могут терять свои характеристики и трансдифференцироваться в фибробласто-подобные клетки. Хотя общая значимость EndMT в основных кардиальных патологиях остается предметом дискуссий, этот процесс может вносить дополнительный вклад в популяцию фибробластов и усиливать фиброз.
Понимание этих фундаментальных механизмов межклеточного взаимодействия является основой для разработки новых терапевтических подходов, нацеленных на модуляцию фибротического ответа.
5.0 Анализ Терапевтических Стратегий, Направленных на Фибробласты
Неэффективность существующих неспецифических методов лечения стимулировала разработку инновационных подходов, нацеленных непосредственно на патологически активированные фибробласты. Эти стратегии основаны на глубоком понимании клеточной и молекулярной биологии фиброза и стремятся обеспечить высокую селективность воздействия.
5.1 Ограничения Существующих Подходов и Необходимость Таргетной Терапии
Многие глобальные ингибиторы, которые демонстрировали эффективность в доклинических моделях, потерпели неудачу в клинических испытаниях. Ярким примером являются ингибиторы p38 MAPK. Несмотря на то, что специфическая блокировка этого пути в фибробластах эффективно подавляет фиброз, системное применение малых молекул-ингибиторов p38 оказалось неэффективным. Вероятно, это связано с повсеместной природой стрессового ответа, опосредованного p38, во многих типах кардиальных клеток, где он выполняет важные физиологические функции, что и подчеркивает критическую необходимость разработки методов лечения с высокой селективностью в отношении именно фибробластов.
5.2 Генная Терапия на Основе Аденоассоциированного Вируса (AAV)
AAV-терапия представляет собой перспективный инструмент для таргетной доставки терапевтических генов в фибробласты сердца.
• Эффективные серотипы: В ходе скрининга было установлено, что серотипы AAV9 и химерный AAV-DJ наиболее эффективно трансдуцируют сердечные фибробласты in vivo.
• Обеспечение специфичности: Для достижения целенаправленной экспрессии гена в фибробластах используются промоторы генов, которые активны именно в этих клетках. Например, промотор гена Tcf21 позволяет воздействовать на всю популяцию фибробластов, тогда как промотор гена Postn активен только в активированных миофибробластах, что открывает возможность для воздействия исключительно на патологические клетки.
5.3 CAR-T-клеточная Терапия для Элиминации Фибробластов
Инновационная стратегия, заимствованная из онкологии, заключается в использовании Т-клеток с химерным антигенным рецептором (CAR-T) для целенаправленного уничтожения фибробластов.
• Ключевая мишень: В качестве мишени был выбран Белок Активации Фибробластов (FAP), который экспрессируется на поверхности патологически активированных фибробластов, но практически отсутствует в здоровых тканях.
• Два подхода к реализации:
1. Адоптивный перенос: Пациенту вводятся предварительно генно-инженерные CAR-T-клетки, нацеленные на FAP.
2. Репрограммирование in vivo: Более современный подход, при котором собственные Т-клетки пациента репрограммируются непосредственно в организме с помощью липидных наночастиц, доставляющих мРНК, кодирующую CAR. Преимущество этого метода заключается во временном эффекте (мРНК со временем деградирует), что потенциально делает терапию более безопасной по сравнению с постоянной экспрессией CAR при адоптивном переносе.
5.4 Механотерапевтические Подходы
Основываясь на понимании механочувствительных путей как ключевых драйверов самоподдерживающегося фиброза (см. раздел 3.3), возникло новое терапевтическое направление, нацеленное на разрыв порочного круга механотрансдукции.
• Ингибирование пути YAP/TAZ: Специфическая делеция YAP/TAZ в фибробластах показала способность уменьшать фиброз после инфаркта миокарда, что делает этот путь привлекательной мишенью.
• Двойное ингибирование: Комплексный подход, сочетающий подавление Src-зависимого механовосприятия и сигналинга TGF-β, продемонстрировал улучшение сократительной функции в мышиной модели сердечной недостаточности. Это подчеркивает потенциал комбинированных механотерапевтических стратегий.
6.0 Заключение и Перспективы Развития
Проведенный анализ демонстрирует парадигмальный сдвиг в подходе к лечению фиброза миокарда: от рассмотрения фиброза как монолитного конечного результата к его восприятию как динамического, управляемого клеточными состояниями процесса. Именно это новое понимание делает концептуально возможными такие инновационные подходы, как генная терапия на основе AAV и клеточная терапия CAR-T, нацеленные на селективную модуляцию или элиминацию патологически активированных фибробластов.
Решающую роль в этом прогрессе сыграли передовые технологии, в частности, модели отслеживания клеточных линий (lineage tracing) и секвенирование РНК единичных клеток (scRNA/snRNA-seq). Эти методы позволили не только детально охарактеризовать гетерогенность фибробластов, но и выявить новые терапевтические мишени и понять сложные механизмы межклеточных взаимодействий в фибротическом микроокружении.
Ключевой задачей на будущее является разработка и внедрение в клиническую практику неинвазивных биомаркеров и методов визуализации, специфичных для фибробластов. Такие инструменты необходимы для мониторинга эффективности антифибротической терапии и оценки обратимости фиброза в реальном времени. Способность точно определять, является ли фиброз у конкретного пациента обратимым или перманентным, будет иметь решающее значение для прогнозирования исходов сердечной недостаточности и персонализации лечения, включая такие состояния, как ремиссия, восстановление или прогрессирование заболевания.