В первой части публикации в отношении пострегистрационных изменений лекарственных препаратов для перорального применения с системным действием обсудили изменения состава вспомогательных веществ и основные требования, которые применяются к оценке степени изменений для выбора оптимальной стратегии исследований, результаты которых будет необходимо представить регулятору.
В этой публикации освещены пострегистрационные изменения места и технологии производства, укрупнение (масштабирование) серий и замена или включение новой АФС (активной фармацевтической субстанции).
1 Изменение места производства
1.1 Документы Евразийского экономического союза (ЕАЭС)
Согласно Приложению №19, Решения №78 Совета Евразийской экономической комиссии (ЕЭК), изменения касающиеся места производства относятся к коду изменений – Б.II.б.1. Добавление или замена производственной площадки относится к изменению Б.II.б.1.д, процедура изменения типа - IB.
Среди необходимых условий, а также документов и данных не указана необходимость предоставления результатов ТСКР или обоснования непредоставления результатов нового исследования биоэквивалентности в соответствии с Правилами проведения исследований биоэквивалентности Союза (Решение №85 Совета ЕЭК).
За исключением п. 5 «Данные анализа одной промышленной серии и двух опытно-промышленных серий, имитирующих процесс производства (или две промышленные серии) и сравнительные данные с тремя сериями, произведенными на предыдущей производственной площадке…», который можно трактовать как необходимость выполнения сравнительного растворения одной промышленной серии и двух опытно-промышленных серий, имитирующих новый процесс производства (или две промышленные серии) с тремя сериями произведенными на предыдущей производственной площадке.
Учитывая, что речь идет, по сути, о данных сертификата анализа, то требуется испытание «Растворение» в рамках собственных/фармакопейных требований к высвобождению, т. е. в среде контроля качества, согласно условиям описанным в соответствующей спецификации по качеству.
В разделе VI Решения №85 Совета ЕЭК и Приложении №5 Решения №85 Совета ЕЭК указания по необходимости предоставления результатов ТСКР или исследований биоэквивалентности при изменении места производства отсутствуют.
1.2 Зарубежные регуляторные документы
В руководстве FDA для изменений[1], связанных с изменением места производства, выделяют 3 уровня степени изменений:
Изменения уровня 1 представляют собой изменения внутри одной площадки.
Изменения уровня 2 представляют собой изменения внутри сопряженных зданий или между площадками, располагающимися в соседних зданиях.
Изменения уровня 3 представляют собой перенос производства в другое здание. Под другим зданием понимается здание, не располагающееся в составе сопряженных зданий, или не располагающиеся вблизи друг к другу здания.
При этом неизменными должны быть: технология производства, а также оборудование, стандартные операционные процедуры (СОПы), условия окружающей среды (например, температура и влажность), контроль и персонал, и отсутствие изменений в протоколах промышленных серий, за исключением административных изменений адреса площадки (например, переименования улицы или номера здания). Под неизменным персоналом понимается квалификация работников, работающих в здании и обладающих необходимым опытом осуществления процесса производства.
Требуемая документация по растворению
Для изменений 1 и 2 уровня, помимо собственных/фармакопейных требований к высвобождению не требуется.
Для изменений 3 уровня - многоточечный профиль растворения в фармакопейной среде или среде заявителя на 15, 30, 45, 60 и 120 минутах или до достижения асимптоты предлагаемой и ныне одобренной лекарственных форм.
Документация по изучению биоэквивалентности in vivo - не требуется для любого уровня изменений.
[1] https://www.fda.gov/media/70949/download.
1.3 Позиция ФГБУ НЦЭСМП Минздрава России
В официальном телеграмм канале ФГБУ НЦЭСМП Минздрава России указано на необходимость предоставления результатов ТСКР при смене или добавлении производственной площадки[2][3][4]:
При замене или добавлении новой производственной площадки для небиологических ЛП для приема внутрь при неизменности технологии произвосдтва
1 «Необходимо предоставление результатов ТСКР в 3-х средах растворения + в среде нормативной документации (НД), 3-х серий (1 серия со «старой» и 2 серии с «новой» производственных площадок) для подтверждения постоянства производства (приложение 5 к 85 Решению)».
В самом Приложении №5 к Решению 85 требований по количеству серий и количеству сред нет.
2 «Также согласно пункту 37 Рекомендации Коллегии ЕЭК от 29 января 2019 г. N 3 «О Руководстве по производству готовых лекарственных форм лекарственных препаратов» независимо от числа производственных площадок, участвующих в процессе производства лекарственного препарата, для производства этого лекарственного препарата следует использовать единый процесс производства.
Таким образом для ЛП, произведенного на разных площадках (по одной технологии), должны быть предоставлены данные ТСКР на образцах двух серий с вводимой площадки и 1 серии со старой площадки».
3 «В случае замены или добавлении новой производственной площадки (код Б.II.б.1.д «Площадка, на которой осуществляются любые производственные операции для нестерильных лекарственных препаратов, за исключением выпуска серий, контроля серий, первичной и вторичной упаковки») необходимо в составе регистрационного досье представить тест сравнительной кинетики растворения (ТСКР) серий ЛП с новой и старой производственной площадки».
При этом в документах для кода Б.II.б.1.д ТСКР не указан.
[2] https://t.me/fgbuexpmed/603.
[3] https://t.me/fgbuexpmed/426.
[4] https://t.me/fgbuexpmed/374.
Заключение
При изменении места производства выполнение ТСКР конечно требуется, но в документах ЕАЭС напрямую не указано, что ТСКР должен выполняться в 3 средах+среде НД для 3-х серий (1 серия со «старой» и 2 серии с «новой» производственных площадок).
По логике документов ЕАЭС (Решение №78) и зарубежных документов достаточно выполнение ТСКР в 1 среде НД. В отношении количества серий следует ориентироваться на 2 промышленные серии с «новой» площадки (или 1 промышленная + 2 опытно-промышленные серии) и 3 серии со «старой» площадки.
Но как указано в телеграмм канале ФГБУ НЦЭСМП Минздрава России «В целях исключения рисков обидных отказов, пожалуйста, НЕ забывайте, что пока еще не для всех типов изменений возможен запрос, в том числе, СТКР для вышеописанных случаев». Поэтому чтобы избежать этих рисков, лучше выполнить предъявляемые требования т.к. возможности обосновать иную позицию может не быть.
2 Документы ЕАЭСлогии/процесса производства
1.1 Документы ЕАЭС
Согласно Приложению №19, Решения №78 Совета ЕЭК, изменения касающиеся технологии производства относятся к коду изменений – Б.II.б.3. Даже при незначимых изменениях производственного процесса Б.II.б.3.а (относится к изменению типа IА) необходимо выполнить ТСКР. Необходимо изучить одну промышленную серию, произведенную по новой технологии в сравнении с 3 сериями, произведенными с помощью предыдущего процесса (п. 3 раздела «документы»).
Также при незначимых изменениях необходимо обоснование непредоставления результатов нового исследования биоэквивалентности в соответствии с Правилами проведения исследований биоэквивалентности Союза (Решение №85 Совета ЕЭК) (п. 4 раздела «Документы»).
Пункт 122, раздела VI Решения №85 Совета ЕЭК говорит то том, что «Всякое представленное обоснование должно основываться на общих принципах, в частности, указанных в приложении N 4 к настоящим Правилам, или при установлении приемлемой (уровня A) in vitro - in vivo корреляции (IVIVC)».
Т.е. требуется предоставление документов обосновывающих биовейвер основанный на биофармацевтической системе классификации (БСК) или предоставлять обоснование биовейвера на основании имеющейся in vitro - in vivo корреляции (IVIVC) уровня А.
При этом стоит учитывать, что для препаратов с немедленным высвобождением биовейвер по БСК затрагивает только препараты с АФС I и III класса по БСК. И таким образом для препаратов с АФС II и IV класса требуется наличие IVIVC уровня А.
2.2 Зарубежные документы
Для научного обоснования отсутствия необходимости в проведении нового исследования БЭ возможно использовать также зарубежные регуляторные и научные подходы, в частности руководство для промышленности FDA [1].
Для изменений технологического процесса руководство выделяет 3 уровня степени изменений:
Изменения уровня 1
Определение
Настоящая категория представляет собой изменения технологического процесса, включая такие изменения, как: изменение времени перемешивания (mixing times) и скорости выполнения операций, укладывающиеся в диапазоны, указанные в досье/диапазон валидации.
Документация по результатам испытаний
Документация по растворению
Помимо собственных/фармакопейных требований к высвобождению не требуется.
Документация по изучению биоэквивалентности in vivo
Не требуется.
Изменения уровня 2
Определение уровня
Настоящая категория представляет собой изменения технологического процесса, включая такие изменения, как: изменение времени перемешивания (mixing times) и скорости выполнения операций, не укладывающиеся диапазоны, указанные в досье/диапазон валидации.
Документация по результатам испытаний
Документация по растворению
Многоточечный профиль растворения в собственной/фармакопейной среде на 15, 30, 45, 60 и 120 минутах или до достижения асимптоты предлагаемой и ныне одобренной лекарственных форм
Документация по изучению биоэквивалентности in vivo
Не требуется.
Изменения уровня 3
Определение уровня
Настоящая категория представляет собой изменения вида технологического процесса производства лекарственного препарата, например, изменение с влажной грануляции на прямое прессование сухого порошка.
Документация по результатам испытаний
Документация по растворению
Многоточечный профиль растворения в собственной/фармакопейной среде на 15, 30, 45, 60 и 120 минутах или до достижения асимптоты предлагаемой и ныне одобренной лекарственных форм
Документация по изучению биоэквивалентности in vivo
Требуется исследование биоэквивалентности in vivo. Исследование биоэквивалентности допускается не проводить, если установлена должная in vivo/in vitro корреляция.
Таким образом, не для всех изменений технологии производства препарата требуется выполнение нового исследования биоэквивалентности.
2.3 Позиция ФГБУ НЦЭСМП Минздрава России
До настоящего времени официальные публикации с разъяснениями этого вопроса отсутствуют.
Заключение
Согласно документам ЕАЭС (Решение №78) даже при незначительных изменениях требуется выполнить ТСКР и обосновать отсутствие необходимости выполнения нового исследования биоэквивалентности.
ТСКР следует выполнить для одной промышленной серии, произведенной по новой технологии в сравнении с 3 сериями, произведенными с помощью предыдущего процесса. Требования по средам растворения не указаны, поэтому следует выполнить ТСКР в среде контроля качества, как это предлагает руководство FDA для аналогичных изменений.
В отношении обоснования отсутствия необходимости выполнения нового исследования биоэквивалентности - если изменения технологии укладываются в уровни 1 и 2 описанные в руководстве FDA, то для обоснования непроведения нового исследования возможно использовать соответствующие критерии и документы по растворению, т.к. изменения 1 и 2 уровня не могут привести к значимому изменению биодоступности лекарственного препарата.
В случае неэквивалентности профилей растворения или в случае изменений уровня 3 по критериями руководства FDA потребуется новое исследование биоэквивалентности. Однако возможен биовейвер основанный на БСК для препаратов с АФС I и III класса, или биовейвер основанный на установленной IVIVC уровня А для препаратов с АФС II и IV класса.
3 Изменение размера серий
3.1 Документы ЕАЭС
Согласно Приложению №19, Решения № 78 Совета ЕЭК, изменения касающиеся укрупнения или разукрупнения размеров серий относятся к коду изменений – Б.II.б.4. Применительно к препаратам не биологического происхождения выделяют укрупнение серий до 10 раз (Б.II.б.4.а), разукрупнение до 10 раз (Б.II.б.4.б) и укрупнение более 10 раз (Б.II.б.4.д).
Среди необходимых условий, а также документов и данных не указана необходимость предоставления результатов ТСКР или обоснования непредоставления результатов нового исследования биоэквивалентности в соответствии с Правилами проведения исследований биоэквивалентности Союза (Решение №85 Совета ЕЭК).
За исключением документов указанных в п. 2 для укрупнения более 10 раз - «Данные анализа серий (в формате сравнительной таблицы) по меньшей мере одной промышленной серии, произведенной в зарегистрированном и предлагаемом размерах. По запросу необходимо представить данные по следующим двум полным промышленным сериям; держатель РУ обязан сообщить, если результаты анализа не укладываются в спецификацию, и предложить план действий», который можно трактовать как необходимость выполнения растворения одной промышленной серии до масштабирования и одной серии после масштабирования.
Учитывая, что речь идет, по сути, о данных сертификата анализа, то требуется испытание «Растворение» в рамках собственных/фармакопейных требований к высвобождению, т. е. в среде контроля качества, согласно условиям описанным в соответствующей спецификации по качеству. При этом держатель РУ должен уведомлять только если результаты не укладываются в спецификацию.
В разделе VI Решения №85 Совета ЕЭК и Приложении №5 Решения №85 Совета ЕЭК указания по необходимости предоставления результатов ТСКР или исследований биоэквивалентности при изменении размера серий отсутствуют.
3.2 Зарубежные регуляторные документы
В руководстве FDA для изменений1, связанных с укрупнением серий также выделяют 2 уровня степени изменений:
Изменения уровня 1
Определение уровня
Изменение размера серии, не превышающее более чем в 10 раз размер серии, использовавшейся в пилотных исследованиях или исследованиях биоэквивалентности при условии, что: 1) оборудование, использованное в производстве опытных серий, имеет ту же конструкцию и функционирует на тех же принципах; 2) серии производятся в полном соответствии текущей надлежащей производственной практикой; 3) в производстве опытных и промышленных серий использовались одинаковые стандартные операционные процедуры (СОПы) и контроли, а также процедуры приготовления и производства.
Документация по результатам испытаний
Документация по растворению
Помимо собственных/фармакопейных требований к высвобождению не требуется.
То есть требуется не сравнительное испытание растворения согласно соответствующему показателю НД.
Документация по изучению биоэквивалентности in vivo
Не требуется.
Изменения уровня 2
Определение уровня
Изменение размера серии, превышающее более чем в 10 раз размер серии, использовавшейся в пилотных исследованиях или исследованиях биоэквивалентности при условии, что: 1) оборудование, использованное в производстве опытных серий, имеет ту же конструкцию и функционирует на тех же принципах; 2) серии производятся в полном соответствии с текущей надлежащей производственной практикой; 3) в производстве опытных серий и промышленных серий использовались одинаковые СОПы и контроли, а также процедуры приготовления и производства.
Документация по результатам испытаний
Документация по растворению
Многоточечный профиль растворения в собственной/фармакопейной среде на 15, 30, 45, 60 и 120 минутах или до достижения асимптоты предлагаемой и ныне одобренной лекарственных форм.
Документация по изучению биоэквивалентности in vivo
Не требуется.
Таким образом, многоточечный ТСКР в среде контроля качества требуется только при увеличении размера серий более чем в 10 раз.
3.3 Позиция ФГБУ НЦЭСМП Минздрава России
До настоящего времени официальные публикации с разъяснениями этого вопроса отсутствуют.
Заключение
Согласно документам ЕАЭС (Решения №78, 85) указания о необходимости выполнения ТСКР и предоставления обоснования отсутствия необходимости выполнения нового исследования биоэквивалентности отсутствуют. Имеется только указание на сравнение серий по данным анализа, т.е. среди прочего по показателю «Растворение» в рамках нормы указанной в НД (одноточечное испытание растворения). Растворение следует выполнить для одной промышленной серии до масштабирования и одной серии после масштабирования в среде контроля качества.
Руководство FDA рекомендует выполнять многоточечный ТСКР для укрупнения более чем в 10 раз.
Поэтому можно рекомендовать помимо испытания «Растворение» по НД также выполнить многоточечный ТСКР в среде контроля качества, как это предлагает руководство FDA для аналогичных изменений.
4 Изменения касающиеся замены или включения новой АФС
4.1 Документы ЕАЭС
Согласно Приложению №19, Решения 78 Совета ЕЭК, изменения касающиеся замены или включения новой АФС для небиологических препаратов относятся к коду изменений – Б.I.а.1, а именно к кодам:
Б.I.а.1.а. – «предлагаемый производитель принадлежит к той же фармацевтической группе, что и одобренный производитель»;
Б.I.а.1.б – «внесение нового производителя активной фармацевтической субстанции, обоснованной МФАФС»;
Б.I.а.1.в - предлагаемый производитель использует резко отличающийся способ синтеза или условия производства, которые могут изменить важные показатели качества активной фармацевтической субстанции, такие как качественный и (или) количественный профиль примесей, требующий квалификации, или физико-химические свойства, влияющие на биодоступность;
Б.I.а.1.ж – «внесение нового производителя активной фармацевтической субстанции, не имеющей МФАФС и требующей существенного обновления соответствующего раздела досье по активной фармацевтической субстанции».
За исключением кода Б.I.а.1.а. остальные коды предусматривают процедуру значимых изменений II типа при этом среди необходимых условий, а также документов и данных не указана необходимость предоставления результатов ТСКР или обоснования непредоставления результатов нового исследования биоэквивалентности в соответствии с Правилами проведения исследований биоэквивалентности Союза (Решение №85 Совета ЕЭК).
Среди необходимых документов в п.4 указано «Данные анализа серий (в формате сравнительной таблицы) по меньшей мере двух серий (по меньшей мере, опытно-промышленных) активной фармацевтической субстанции от текущих и предлагаемых производителей (площадок)». Что можно трактовать как необходимость выполнения растворения одной серии, произведённой из старой субстанции и двух серий, произведённых из новых АФС. Учитывая, что речь идет, по сути, о данных сертификата анализа, то требуется испытание «Растворение» в рамках собственных/фармакопейных требований к высвобождению, т.е. в среде контроля качества, согласно условиям описанным в соответствующей спецификации по качеству.
В разделе VI Решения №85 Совета ЕЭК и Приложении №5 Решения №85 Совета ЕЭК указания по необходимости предоставления результатов ТСКР или исследований биоэквивалентности при изменении АФС отсутствуют.
4.2 Зарубежные регуляторные документы
В руководстве FDA для изменений, связанных с изменением субстанции указано следующее[5].
Если изменение в производстве лекарственного вещества (АФС) может повлиять на физико-химические свойства или биодоступность лекарственного препарата, эквивалентность этой серии относительно лекарственного препарата, изготовленного из лекарственного вещества (АФС) до модификации, должна быть установлена с помощью соответствующей процедуры испытания in vitro. Тип испытания, который будет подходящим, будет зависеть от лекарственной формы, пути введения и растворимости лекарственного вещества (см. рисунок ниже).
[5] https://www.fda.gov/media/115733/download.
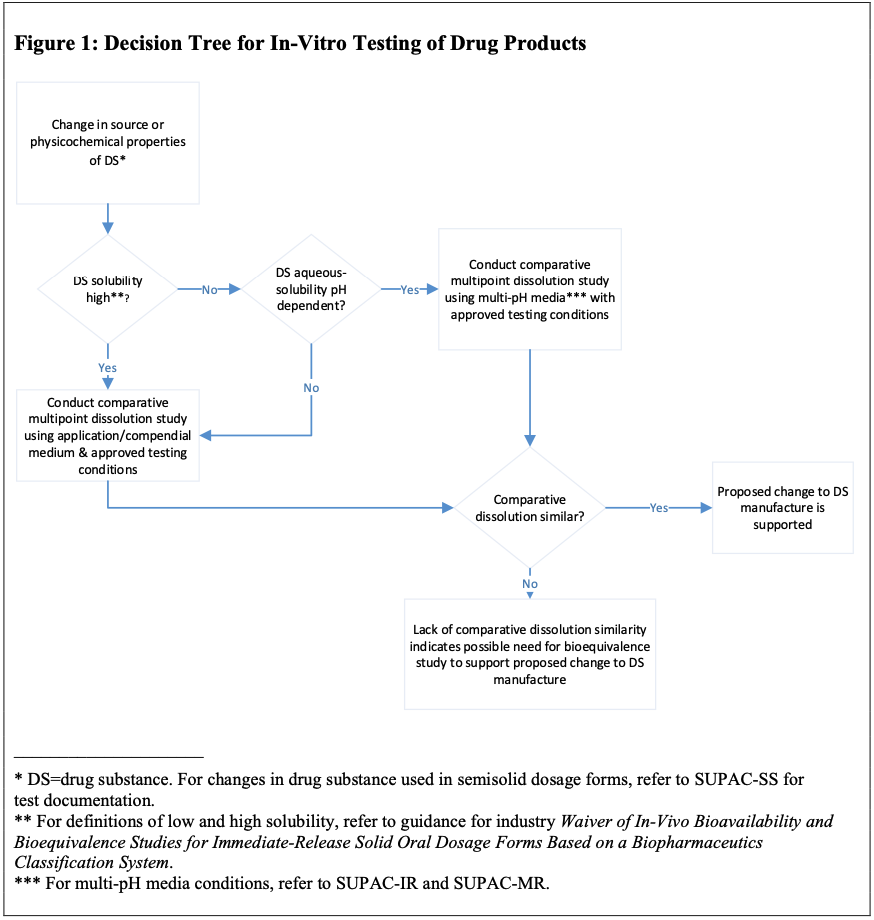
Таким образом, для препаратов с АФС с высокой растворимостью (I и III класс БКС) достаточно выполнения ТСКР в среде контроля качества, для препаратов с низкой растворимостью (II и IV класс БСК), но если растворимость зависит от pH, то требуется выполнения ТСКР в средах с физиологическими значениями pH (1.2, 4.5, 6.8). В случае если ТСКР продемонстрирует неэквивалентность, то потребуется выполнить исследование биоэквивалентности.
4.3 Позиция ФГБУ НЦЭСМП Минздрава России
В официальном телеграмм канале ФГБУ НЦЭСМП Минздрава России указано на необходимость предоставления результатов ТСКР при смене или добавлении АФС [6][7]:
1. Для кода Б.I.а.1.а) – указано: не нужно ТСКР, это изменение без экспертизы. Важно соблюдение условий и правильная классификация изменения.
2. Для кода Б.I.а.1.в) – указано: это изменение II типа. Влияние на биодоступность требует проведения уже клинического исследования БЭ. Т.е. тут необходимо рассмотрение всей совокупности данных и разброс необходимых результатов исследований может быть большой: от теоретического обоснования до исследования БЭ. Именно поэтому это II тип.
3. Для кода Б.I.а.1.и) - указано: ТСКР нужен, нужно подтвердить постоянство производства с изменением площадки, которая может критично повлиять на это.
4. Для кодов Б.I.а.1.б и код Б.I.а.1.ж – указано: необходимо в составе регистрационного досье представить тест сравнительной кинетики растворения (ТСКР) между ЛП со старой и новой субстанцией.
При этом уточнений по выбору сред растворения и количества изучаемых серий не опубликовано.
Согласно запросам ФГБУ НЦЭСМП Минздрава России, предлагается выполнять сравнение одной серии произведенной из старой АФС (промышленная серия) и двух серий произведенных из новой АФС (по меньшей мере, опытно-промышленных серий) в трех средах растворения (pH 1.2, 4.5, 6.8) и среде контроля качества (если отличается от вышеуказанных трех сред).
[6] https://t.me/fgbuexpmed/400.
[7] https://t.me/fgbuexpmed/374.

Заключение
Согласно документам ЕАЭС (Решения №78, 85) указания о необходимости выполнения ТСКР (в 3 средах + среде контроля качества) и предоставления обоснования отсутствия необходимости выполнения нового исследования биоэквивалентности при изменении АФС отсутствуют.
Однако по аналогии с требованиями, которые применяются в отношении изменения места производства, которые в документах ЕАЭС также не прописаны в отношении выбора сред и количества серий, следует ориентироваться на то, что потребуется предоставить сравнение одной серии произведенной из старой АФС (промышленная серия) и двух серий произведенных из новой АФС (по меньшей мере, опытно-промышленных серий) в трех средах растворения (pH 1.2, 4.5, 6.8) и среде контроля качества (если отличается от вышеуказанных трех сред).
При этом вопрос необходимости предоставления результатов в трех средах и среде контроля качества не в полной мере обоснован, т.к. методы испытаний растворения необходимо разработать применительно к каждому лекарственному препарату, и методика должна обладать достаточной дискриминационной способностью, т.е. улавливать разницу между сериями с приемлемой и неприемлемой биодоступностью, и именно на ее основе разрабатывается показатель растворение для НД. Поэтому в большинстве случаев для оценки растворения серий в случае изменений как правило достаточно ТСКР в одной среде контроля качества, особенно если речь идет в отношении контроля качества серий в целях подтверждения постоянства производства. Более того некоторые АФС могут вовсе не растворяться в некоторых из трех «стандартных» сред, особенно если это АФС II или IVкласса по БСК и требуют модификации сред путем добавления поверхностно активных веществ.
Таким образом, самый простой способ — это выполнить требования в отношении трех сред + среды контроля качества, но также можно обосновать и альтернативные подходы, которые допустимы документами ЕАЭС и, в частности, Приложением №5 Решения 85 ЕЭК.
Если возникнут вопросы или сложности в определении степени изменений пишите в комментарии или в канале - https://t.me/bioequivalence_in_EAEU под публикацией.