Новая регуляторная система Евразийского экономического союза (далее Союза) значительно подняла планку требований, предъявляемых к лекарственным препаратам (ЛП) в части документации регистрационного досье. С регистрацией и приведением в соответствие досье все более-менее понятно и данные вопросы достаточно подробно отражены в нормативно правовых актах (НПА) Союза и широко обсуждаются на профильных конференциях и телеграмм каналах.
Вместе с тем вопросы пострегистрационных изменений являются до настоящего времени проблемными. Несмотря на доработку НПА Союза по этой теме вопросы классификации изменений и объем требуемой документации остаются зачастую открытыми, в связи с чем и эксперты, и представители фармотрасли часто путаются в том, какие результаты каких исследований необходимы.
Чаще всего пострегистрационные изменения связаны с изменением состава вспомогательных веществ (ВВ), поэтому в первой публикации остановимся на изменениях состава ВВ в Союзе.
В НПА Союза однозначной трактовки требований по объему предоставляемых данных нет, но есть ключевые НПА, на которые можно опираться.
- В Правилах регистрации Союза (Решение №78) в Приложении 19 для внесения изменений в состав ВВ в рубрике "Б.II.а.3" приводятся требуемые условия и документы (достаточно поверхностно и с техническими ошибками).
- В Рекомендации Коллегии ЕЭК №28 от 10.09.2019 в Разделе III, пп. 14-16 и Приложении №2 представлены критерии оценки значимости изменения состава ВВ и пример расчета с отсылкой на Приложение 4 к правилам исследований биоэквивалентности (БЭ) (Решение №85) (не значимые изменения - не превышающие +/-5%).
- В Решении 85 есть пункт 122, в котором сказано:
«При изменении ранее одобренного состава … которые могут повлиять на биодоступность, проводится исследование биоэквивалентности, если не представлено иных обоснований. Всякое представленное обоснование должно основываться на общих принципах, в частности указанных в Приложении 4 к правилам». И соответственно есть Приложение №4 (Требования к биовейверу на основании БСК) в котором указано, что он может применяться при внесении изменений в досье, требующих установления биоэквивалентности.
Ориентируясь на эти НПА, можно судить:
1. Что незначимые изменения ВВ точно не требующие исследований биоэквивалентности - это изменения не превышающие в сумме +/-5%, и в качестве условия, требующие чтобы профиль растворения был сопоставим для серий нового состава в сравнении со старым составом (для 2 серий). При этом в качестве предоставляемого документа ТСКР не значится (в Приложении 19 Решения №78).
2. Изменения связанные с заменой ВВ на аналогичное ВВ требуют ТСКР (для серий нового состава в сравнении со старым составом (для 2 серий)) и обоснования непредставления результатов нового исследования биоэквивалентности.
3. Все прочие изменения по умолчанию должны рассматриваться, как изменения, требующие исследования биоэквивалентности или обоснования непроведения БЭ (п. 122 Правил БЭ), опираясь на способность ВВ повлиять на биодоступность.
Таким образом, все изменения состава ВВ более +/-5%* (в сумме или превышающие значения, указанные в Приложении №4 к Правилам БЭ) требуют предоставления не только ТСКР, но и как минимум данных в поддержку, что изменения не влияют на биодоступность и нового исследования биоэквивалентности не потребуется.
* - после вступления в силу поправок в Решение №85 (Совет Евразийской экономической комиссии 12 апреля внес изменения в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза) в Приложение №4, норма 5% после 12 мая будет изменена на 10%. При том что норма значимости изменений останется на уровне 5% согласно Рекомендации Коллегии ЕЭК №28.
При этом в НПА Союза нет документов, которые определяли, бы какие изменения могут оказывать влияние на биодоступность. Соответственно эта задача возлагается полностью на Заявителей.
Однако есть НПА других стран, где требования к регулированию обращения лекарств не ниже, а даже выше, чем установленные в Союзе. Например, в США FDA еще в 1995 году выпустило руководство: Guidance for Industry: SUPAC-IR Immediate Release Solid Oral Dosage Forms, Scale - Up and Post-approval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, In Vivo Bioequivalence Documentation, Guidance November 1995.
В данном Руководстве отражено представление о степени значимости изменений состава ЛП и описан объем данных, необходимых к предоставлению на экспертизу регулятору обращения ЛП.
Ознакомиться с регуляторными требования в отношении пострегистрационных изменений можно в публикации - http://www.fptl.ru/biblioteka/razrabotka-i-ekspertiza-LS/NCJeSMP_2014-03_STKR-pri-postregistracionnih-ismeneniyah.pdf.
В таблицах ниже приведены градации степени изменений вспомогательных веществ ЛП с немедленным высвобождением (НВ) согласно SUPAC-IR.
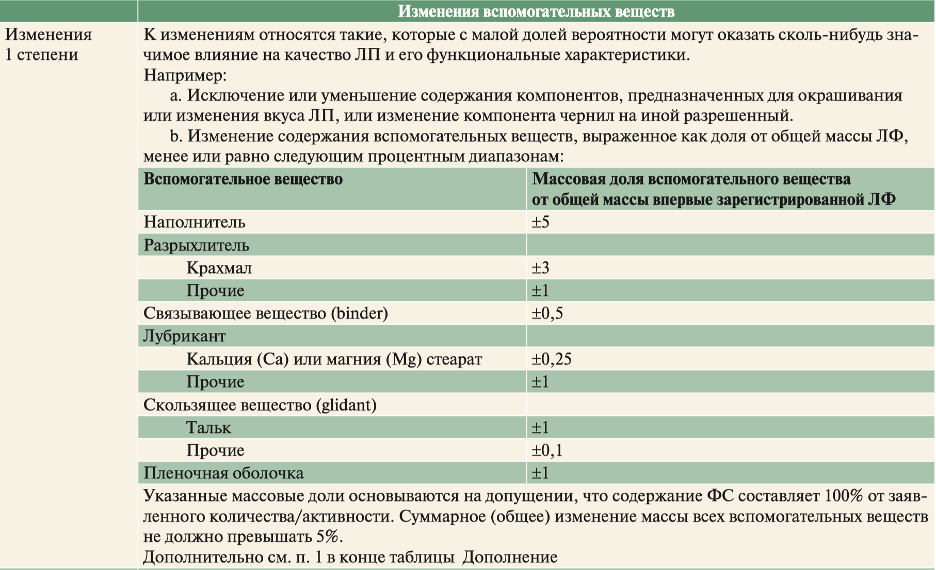
Пример, в лекарственном препарате, состоящем из фармацевтической субстанции А, лактозы, микрокристаллической целлюлозы и магния стеарата, в целях соблюдения критериев уровня 1 совокупное содержание лактозы и микрокристаллической целлюлозы не должно изменяться более чем на 5 % от общей абсолютной массы (например, содержание лактозы увеличивается на 2,5 %, а микрокристаллической целлюлозы снижается на 2,5 %) относительно массы впервые зарегистрированной лекарственной формы.
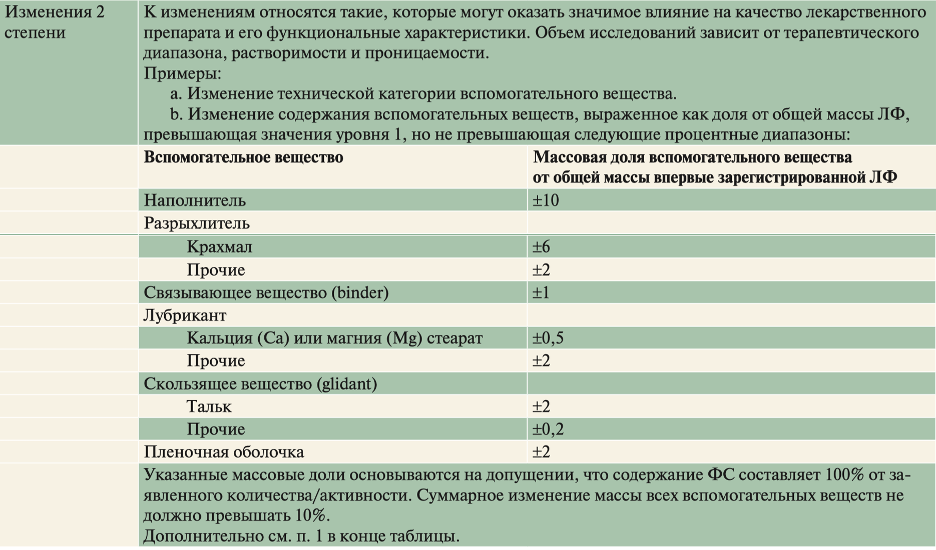
Пример 1, изменение технической категории вспомогательного вещества с авицела PH102 на авицел PH200.
Пример 2, в лекарственном препарате, состоящем из фармацевтической субстанции А, лактозы, микрокристаллической целлюлозы и магния стеарата, в целях соблюдения критериев уровня 2 совокупное содержание лактозы и микрокристаллической целлюлозы не должно изменяться более чем на 10 % от общей абсолютной массы (например, содержание лактозы увеличивается на 5 %, а микрокристаллической целлюлозы снижается на 5 %) относительно массы впервые зарегистрированной лекарственной формы.
Как видно из представленной таблицы эти нормы были взяты за основу и в документе ICH M9 и в обновленном Приложении №4 к решению №85 (редакция от 12.04.2024) г. в отношении биовейвера основанного на Биофармацевтической системе классификации (БСК).
Согласно выше указанному документу, требования в отношении предоставляемой документации следующие:
В случае изменений 1 степени достаточно испытания «тест Растворение» согласно собственным фармакопейным требованиям, спецификациям (в условиях из НД).
Исследование БЭ не требуется.
В случае изменений состава 2 степени требуемый тип СТКР определяется в зависимости от класса БСК:
для I класса - растворение 85% за 15 минут в 900 мл 0,1N HCL (тип А СТКР);
для II класса в воде, 0,1 N HCl и буферных средах при pH 4,5; 6,5 и 7,5 (пять профилей) с отбором проб на 15, 30, 45, 60 и 120 минутах до растворения 90% лекарственного средства или до достижения асимптоты (тип С СТКР);
для III класса – в собственной/фармакопейной среде на 15, 30, 45, 60 и 120 минутах или до достижения асимптоты) (тип В СТКР);
для других изменений 2 степени СТКР – как в случае действующего вещества из III класса БСК или согласно собственным фармакопейным требованиям.
Исследование БЭ не требуется.
Для изменений 3 степени требуется новое исследование БЭ или биовейвер основанный на in vivo/in vitroкорреляции и СТКР как в случае действующего вещества из III класса БСК или согласно собственным фармакопейным требованиям.
Применительно к реалиям ЕАЭС эти требования будут выглядеть следующим образом
Типы СТКР при внесении изменений в состав ЛП в зависимости от класса БКС:
1 класс БСК - растворение 85% за 15 минут в средах с pH 1.2, 4.5, 6.8 или фактор f2* более 50 в средах с pH 1.2, 4.5, 6.8 при растворении 85% за 30 минут. Временные точки определения концентрации – например, 10, 15, 30 минут.
3 класс БСК - растворение 85% за 15 минут в средах с pH 1.2, 4.5, 6.8. Временные точки определения концентрации – например, 10, 15, 30 минут.
2 и 4 класс - многоточечный профиль растворения в средах растворения в средах с pH 1.2, 4.5, 6.8 и фармакопейной среде (если отличается). Временные точки определения концентрации – например, 15, 30, 45, 60 и 120 минут или до выхода в раствор до 90% активного вещества средства из ЛП или до достижения предела растворения. Возможно добавление солюбилизаторов - поверхностно активных веществ (ПАВ). Фактор f2* более 50 во всех средах.
* При невозможности применения f2 могут использоваться другие методы сравнения [подробнее смотри - https://doi.org/10.33380/2305-2066-2021-10-4-197-207].
В остальном должны соблюдаться требования к проведению СТКР описанные в Решении №85 (количество изучаемых серий, условия выполнения, аппараты, критерии оценки).
Если возникнут вопросы или сложности в определении степени изменений состава пишите в канал - https://t.me/bioequivalence_in_EAEU