Эндотелий представляет собой один слой эпителия, покрывающий поверхность сосудистой системы, и представляет собой физический барьер между кровью и стенкой сосуда, играющий важную роль в поддержании внутрисосудистого гомеостаза.
Однако эндотелиальная дисфункция или гибель эндотелиальных клеток могут вызывать нарушение сосудистого барьера, вазоконстрикцию и диастолическую дисфункцию, пролиферацию и миграцию гладкомышечных клеток сосудов, воспалительные реакции и тромбозы, которые тесно связаны с прогрессированием некоторых заболеваний, таких как атеросклероз, гипертония, ишемическая болезнь сердца, ишемический инсульт, острое повреждение легких, острое повреждение почек, диабетическая ретинопатия и болезнь Альцгеймера.
Окислительный стресс, вызванный перепроизводством активных форм кислорода (АФК), является важным механизмом, лежащим в основе гибели эндотелиальных клеток.
Все больше данных свидетельствует о том, что АФК могут вызывать гибель эндотелиальных клеток различными способами, включая пироптоз, партанатоз и ферроптоз.
Таким образом, этот обзор будет систематически иллюстрировать источник АФК в эндотелиальных клетках (ЭК); выявить молекулярный механизм, с помощью которого АФК запускают пироптоз, партанатоз и ферроптоз в ЭК; и предоставить новые идеи для исследования и лечения заболеваний, связанных с эндотелиальной дисфункцией.
Эндотелий — высокоактивный монослой эпителия, покрывающий поверхность кровеносных сосудов.
Эндотелий играет важную роль в поддержании вазомоторной, коагуляционной и антикоагулянтной систем, иммунной регуляции, пролиферации и миграции гладких мышц сосудов (1–3).
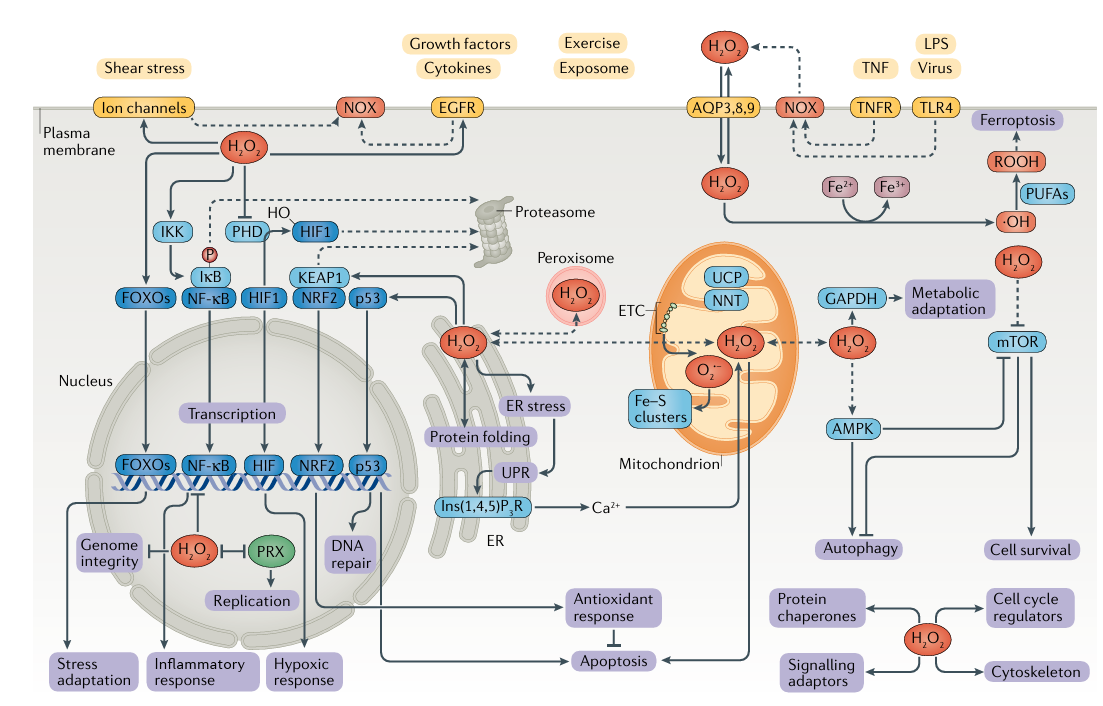
Активные формы кислорода (АФК) в эндотелиальных клетках (ЭК) в основном происходят из митохондрий, НАДФН-оксидазы (NOX), разобщающей eNOS и ксантиноксидазы (ХО) (4, 5).
В процессе АФК-опосредованной ЭД происходит экспрессия различных провоспалительных цитокинов, т.е. интерлейкина-1β (интерлейкин-1β), интерлейкина-18 (интерлейкин-18, IL-18), а также молекул клеточной адгезии, т.е. межклеточных Молекула адгезии-1 (ICAM-1), молекула адгезии сосудистых клеток-1 (VCAM-1) и Е-селектин могут стимулироваться в эндотелиальных клетках. Эти молекулы тесно связаны с возникновением воспалительных реакций (8, 9).
Кроме того, АФК могут опосредовать различные виды запрограммированной гибели клеток (ПКС) в эндотелиальных клетках, такие как пироптоз, партанатоз и ферроптоз.
Стоит отметить, что эндотелиальная дисфункция или гибель эндотелиальных клеток тесно связана с возникновением и развитием различных заболеваний, таких как атеросклероз (10), ишемическая болезнь сердца (11), гипертоническая болезнь (12), ишемический инсульт (13), острый легочный травма (14), острое повреждение почек (15), диабетическая ретинопатия (16) и болезнь Альцгеймера (17) (рис. 1).
Этот обзор систематически раскрывает источники АФК в ЕС; освещает молекулярные механизмы пироптоза, партанатоза и ферроптоза, индуцированного АФК, в клетках ЕС; и предоставить новые идеи для исследования и лечения заболеваний, связанных со смертью эндотелиальных клеток.
Источники АФК
Внутриклеточные АФК в основном состоят из супероксидных анионов (O∙–2), перекиси водорода (H2O2) и гидроксильных радикалов (OH•) (18).
O2 образует O∙–2 путем захвата электрона, что приводит к образованию других АФК. O∙–2 нестабилен в водных растворах из-за короткого периода полураспада; следовательно, внутриклеточный O∙–2 быстро удаляется или преобразуется в другие формы АФК.
O∙–2 выводится или преобразуется в основном тремя путями:
1) O∙–2 генерирует H2O2 под действием супероксиддисмутазы (СОД);
2) низкие концентрации (пикомолярный диапазон) O∙–2 взаимодействуют с оксидом азота (NO) с образованием пероксинитрит-аниона (ONOO•), что происходит даже быстрее, чем диспропорционирование с образованием H2O2;
3) высокие концентрации O∙–2 генерируют OH• в результате реакции Фентона с H2O2 (18).
Кроме того, OH• реагирует с жирными кислотами с образованием свободных радикалов липидов (L•).
АФК в ЭК в основном образуются из митохондрий, НАДФН-оксидазы (NOX), разобщения эндотелиальной NOS (eNOS) и ксантиноксидазы (XO) (4, 5) (рис. 2).
Митохондрии
Митохондрии являются источником клеточной энергии и производят АТФ посредством окислительного фосфорилирования (OXPHO), на которое приходится примерно 80% энергетических потребностей, а на гликолиз приходится оставшиеся 20%.
Производство митохондриальных АФК является результатом окислительного фосфорилирования, связанного с аэробным дыханием в митохондриальной цепи переноса электронов (ETC).
Митохондриальные комплексы I и III являются основными местами генерации O∙–2 (19–21).
Утечка электронов из ЭТЦ приводит к восстановлению O2 до O∙–2, а не до H2O.
СОД дополнительно диспропорционирует митохондриальный O∙–2 с образованием H2O2. По оценкам, примерно 1-2% O2, поступающего в внеземной земной шар, преобразуется в АФК (22) (рис. 2). Более того, перепроизводство митохондриальных АФК является одной из причин дисфункции ЭК. Например, Рао и др. показали, что нокаут никотинамиднуклеотидтрансгидрогеназы (NNT) приводит к значительному увеличению продукции митохондриальных АФК и активности глутатионпероксидазы, а также снижению активности глутатионредуктазы (23).
НАФДН-оксидаза
Структура НАФДН-оксидаза (NOX) является важным источником АФК в клетках. Семейство NOX включает NOX1, NOX2, NOX3, NOX4, NOX5 и двойные оксидазы (DUOX1 и DUOX2) (22). NOX представляют собой мультитрансмембранные белки, С-концы которых расположены в цитоплазме, и они имеют общие домены, включая шесть консервативных трансмембранных доменов, четыре консервативных гем-связывающих гистидина, домены, связывающие флавинадениндинуклеотид (FAD), и домены, связывающие НАДФН. (24).
NOX, в свою очередь, переносит электроны от НАДФН к ФАД, гемовой группе, а затем к O2, что приводит к образованию O∙–2 и/или H2O2 (25).
2.2.2 Активация NOX при ЭД. Основные подтипы NOX при ЭК включают NOX1, NOX2, NOX4 и NOX5 (25, 26). Каталитическим продуктом NOX1, NOX2 и NOX5 является O∙–2, а каталитическим продуктом NOX4 — H2O2 (рис. 3). Комплексы NOX состоят из каталитических субъединиц (NOX) и регуляторных субъединиц, за исключением NOX5, который состоит только из одной каталитической субъединицы (22). NOX2 является первой изоформой NOX, идентифицированной в ЭК, и представляет собой наиболее широко и глубоко изученную изоформу; поэтому мы сначала обсудим механизм его активации. В условиях покоя NOX2 и p22phox расположены на мембране в виде неактивных комплексов, тогда как субъединицы p40phox, p67phox и p47phox расположены в цитоплазме (22). Для активации NOX2 также требуется небольшая ГТФаза Rac1. Активация Rac1 инициирует NOX2, и Rac1 рекрутируется на мембрану, а затем рекрутирует другие цитозольные компоненты (27). Затем p47phox фосфорилируется протеинкиназой C (PKC) и переносится на мембрану вместе с p67phox и p40phox (28). Затем фосфорилирование p47phox может сочетаться с p22phox для реализации сборки и активации комплекса NOX2 (29). Базальная активность NOX2 в ЭК низкая, хотя она быстро активируется патологическими причинными факторами, такими как гиперлипидемия, гипертония и гипергликемия (30). Сообщалось, что повреждение ЭК на ранних стадиях сосудистых заболеваний опосредовано избытком супероксида NOX2 (31). Подобно NOX2, активация NOX1 требует сборки нескольких субъединиц. Во время активации NOX1 функцию активации p67phox выполняет NOXA1, а организаторскую функцию p47phox выполняет NOXO1 (32, 33). По сравнению с p47phox, NOXO1 не содержит домена автоингибитора; следовательно, комплекс NOX1-NOXO1-NOXA1 обладает высокой базальной активностью (29). Отчеты показали, что сверхэкспрессия эндотелина-1 (ET-1) в ЭК способствует прогрессированию атеросклероза через NOX1 при диабете 1 типа, периваскулярном окислительном стрессе и воспалении (34). Кроме того, NOX1 участвует в разъединении eNOS в EC. Например, Юн и др. обнаружили, что активация NOX1 у мышей с диабетом, индуцированным стрептозотоцином, зависит от p47phox и NOXO1 и опосредует разобщение eNOS. Мыши с нокаутом NOX1 защищены от ЭД (35). NOX4 является наиболее экспрессируемым гомологом NOX в ЭК. По сравнению с NOX1 и NOX2, для активации NOX4 требуется только p22phox и белок 2, взаимодействующий с полимеразой дельта (Poldip2) (30, 36).
Несколько исследований показали, что NOX4 играет важную роль в ЭД. Например, Цзян и др. обнаружили, что нокдаун NOX4 ослабляет выработку легочных АФК у септических мышей, ослабляет редокс-чувствительную активацию пути CaMKII/ERK1/2/MLCK и восстанавливает экспрессию белков плотного соединения ZO-1 и окклюдина для поддержания целостности EC-барьера. (37). Чжао и др. показали, что трет-бутилгидропероксид (t-BHP) индуцирует апоптоз EC через NOX4 (38). Однако есть также сообщения о том, что NOX4 защищает ЭК во время окислительного стресса. Это может быть связано с образованием H2O2 NOX4.
H2O2 считается важным сигнальным промежуточным продуктом из-за его способности избирательно и обратимо окислять реактивные остатки цистеина, тем самым изменяя функцию белков-мишеней, включая фосфатазы, киназы, ионные каналы и факторы транскрипции (39).
В отличие от NOX1, NOX2 и NOX4, активация NOX5 не зависит от других субъединиц. NOX5 содержит N-концевой кальмодулинподобный домен с четырьмя сайтами связывания Ca2+ (EF-руки) (39).
Следовательно, активность NOX5 может напрямую регулироваться изменениями внутриклеточного [Ca2+]. Данные свидетельствуют о том, что NOX5 играет важную роль в ЭД. Сильва и др. обнаружили, что лизофосфатидилхолин стимулирует NOX5-зависимое производство АФК в ЭК посредством притока кальция, что приводит к ЭД (40).
Эльбатрик и др. обнаружили, что сверхэкспрессия NOX5 у мышей вызывает разобщение eNOS, что приводит к ЭД (41).
Следовательно, АФК, полученные из NOX, играют важную роль в опосредовании ЭД.
Ксантиноксидоредуктаза (XOR) существует в двух разных формах: ксантиндегидрогеназа (XDH) и XO, и они представляют собой ферменты, лимитирующие скорость метаболизма пуринов (50).
Обычно XOR существует в клетках в форме XDH. XDH представляет собой гомодимер массой около 300 кДа с четырьмя окислительно-восстановительными центрами в каждой субъединице: молибденовым кофактором (Mo-co), двумя железо-серными (Fe-S) центрами и доменом флавинадениндинуклеотида (FAD) (51). XDH катализирует окисление гипоксантина до ксантина и ксантина до мочевой кислоты в сайте Mo-co, а электроны перемещаются через два центра Fe-S к сайту связывания FAD, где НАД+ восстанавливается до НАДН (51). В физиологических условиях XOR в основном присутствует в ЭК в форме XDH (52).
XDH может расщеплять гипоксантин на мочевую кислоту (53) и восстанавливать нитрит с образованием NO, который помогает регулировать вазодилатацию и артериальное давление (53).
Однако в условиях окислительного стресса АФК могут окислять цистинтиолы на XDH, что приводит к превращению XDH в XO (30, 54).
Основное различие между XO и XDH заключается в их сродстве к окислительному субстрату, при этом XO имеет пониженное сродство к NAD+ и более чем в 11 раз повышенное сродство к O2 (54). Способствуя разложению гипоксантина до мочевой кислоты, XO генерирует O∙–2 за счет одноэлектронного восстановления и H2O2 за счет двухэлектронного восстановления (55).
Отчеты показали, что ЭД, вызванная ХО, тесно связана с его побочными продуктами, включая АФК и мочевую кислоту (56, 57).
Внутриклеточная мочевая кислота может усугублять окислительный стресс в ЭК, тем самым вызывая ЭД (52).
То есть при подагре( "подагрическом состоянии"), когда уровень мочевой кислоты в тканях и клетках высокий и далее он высокий в крови, а значит после почечной фильтрации и в моче и всех биологических жидкостях-синовиальной жидкости суставов, цереброспинальной жидкости, т.е. ликвора) это приводит к повреждению не только напрямую действую на систему гомеостаза , но и через выработку активных форм кислорода.
Таким образом, ХО является важным источником АФК в ЭК и тесно связан с ЭД (рис. 2).
Путь пироптоза
В зависимости от того, требуется ли пироптоз активации каспазы-1, его можно разделить на классический и неклассический воспалительные пути (рис. 5).
Классический воспалительный путь в основном включает сборку и активацию воспалительных процессов, образование поринов, а также быстрое и секрецию IL-1β и IL-18. В частности, внутриклеточные и внеклеточные PAMP или DAMP (например, вирусная дцДНК, бактериальный липополисахарид, внеклеточный АТФ, ох-ЛПНП и кристаллы холестерина) могут способствовать сборке и активации воспалительных сомов. Инфламмасомы активируют прокаспазу-1 путем саморасщепления.
Активированная каспаза-1 распределяет порин GSDMD с образованием зрелого N-GSDMD (70) и различает про-IL-1β и про-IL-18 с образованием зрелых IL-1β и IL-18 (70). По сравнению с классическим воспалительным путем активация неканонического воспалительного пути не требует сборки и активации воспалительной системы.
Липополисахарид, компонент бактериальной клеточной стенки, может активировать каспазу-11 (человеческую) или каспазу-4/5 (мышиную) (71, 72). Активированная каспаза-4/5/11 различает GSDMD с образованием зрелого N-GSDMD (64, 65). Закон N-GSDMD интегрируется в клеточную мембрану с образованием мембранных пор, которые опосредуют пироптоз.
В заключение следует отметить, что стимулирующие факторы, такие как ох-ЛПНП, гипергликемия, никотин и Ang II, могут вызывать увеличение АФК в ЭК.
В эндотелиальных клетках АФК действуют как мост между патологическими стимулами, такими как ox-LDL, гипергликемия, Ang II и никотин, и активацией воспалительной NLRP3. Процесс активации воспаления NLRP3 в ЭК, запускаемый АФК, включает два ключевых этапа: фазы инициации и активации. Фаза инициации относится к индуцированной АФК активации NLRP3, каспазы-1, IL-1β и IL-18 в EC. Фаза активации относится к АФК, способствующим сборке и активации воспаления NLRP3 посредством TXNIP. Активация воспалительной сомы NLRP3 способствует созреванию IL-1β и IL-18 и может опосредовать образование порина N-GSDMD, вызывая тем самым набухание и даже разрыв клеток, что приводит к гибели клеток. Кроме того, образование пор мембраны способствует высвобождению клеточных компонентов, включая IL-1β, IL-18 и HMGB1, которые участвуют в воспалительных реакциях. IL-1β, IL-18 и HMGB1 могут связываться с поверхностью EC посредством IL-1R, IL-18R и TLR соответственно и повышать экспрессию ICAM-1 и VCAM-1 через Myd88/IRAK-1/. Путь TRAF-6/NF-κB. Кроме того, активация воспалительной сомы NLRP3 может нарушать белки плотных соединений между ЭК, что приводит к увеличению проницаемости сосудов (рис. 6).
Партанатос — это тип запрограммированной гибели клеток, который зависит от поли(АДФ-рибосомы) полимеразы 1 (PARP-1) (96, 97). PARP-1 представляет собой АДФ-рибозилтрансферазу, которая переносит АДФ-рибозу от никотинамидадениндинуклеотида (НАД+) к белкам-рецепторам (98, 99). PARP-1 первоначально был описан как фермент-сенсор разрыва ДНК, активируемый одно- и двухцепочечными разрывами ДНК (100). Активация PARP-1, вызванная повреждением ДНК, считается классическим путем активации этого фермента. АФК, ионизирующая радиация и алкилирующие агенты являются частыми причинами фрагментации ДНК (101–103). Активация PARP-1 зависит от степени повреждения ДНК. Однако, когда ДНК сильно повреждена, сверхактивация PARP-1 вызывает накопление поли(АДФ-рибозы) (PAR), процесс, который потребляет большое количество НАД+. НАД+ является прямым субстратом для синтеза ПАР и кофактором во многих окислительно-восстановительных реакциях, таких как цикл трикарбоновых кислот, гликолиз и пентозофосфатный путь (104, 105). Более того, транслокация PAR из ядра в митохондрии вызывает высвобождение фактора, индуцирующего апоптоз (AIF) (106, 107). После того как АИФ покидает митохондрии, он образует в цитоплазме комплекс с фактором, ингибирующим миграцию макрофагов (МИФ). Впоследствии ядерная транслокация комплекса AIF/MIF вызывает конденсацию хроматина и фрагментацию ДНК, что в конечном итоге приводит к гибели клеток (108–110) (Figure 7).
Ферроптоз — железозависимый процесс, включающий запрограммированную гибель клеток. Перекисное окисление липидов, представляющее собой процесс, посредством которого OH• атакует двойные углерод-углеродные связи липидов, особенно полиненасыщенных жирных кислот (ПНЖК) (119), является важным маркером ферроптоза.
Таким образом, производство OH• является ключевым фактором в процессе перекисного окисления липидов. O∙–2 реагирует с H2O2 при катализе Fe2+ с образованием Fe3+, OH• и OH-, что называется реакцией Фентона. Кроме того, O∙–2 реагирует с Fe3+ с образованием Fe2+ в процессе, называемом циклом Габера-Вейсса.
Перекисное окисление липидов можно разделить на три стадии: инициация, распространение и терминация.
На начальной стадии OH• взаимодействует с липидами с образованием углеродцентрированных липидных радикалов (L•). L• реагирует с кислородом с образованием липидного пероксидного радикала (LOO•), который далее генерирует новый L• (растущая фаза) и липидный перекись водорода (LOOH) из другого молекулярного липида. L• и LOOH, образующиеся на стадии размножения, могут быть уничтожены антиоксидантными молекулами мевалонатного пути, такими как коэнзим Q10 (CoQ 10) и витамин Е (VitE) (120–122).
Кроме того, исследования показали, что хелаторы железа, такие как дефероксамин (ДФО) и циклопироксоламин (СРХ), могут ингибировать возникновение ферроптоза, ингибируя перекисное окисление липидов (122–125).
Глутамат, цистин и глицин являются сырьем, используемым для синтеза GSH.
Цистин проникает в клетку через аминокислотную антипортерную систему Xc, которая состоит из субъединицы легкой цепи SLC7A11 и субъединицы тяжелой цепи SLC3A2. Система Xc обменивает цистин на глутамат таким образом, что цистин попадает в клетку и далее восстанавливается до цистеина (128).
Глутаматцистеинлигаза (GCL) катализирует образование γ-глутаматцистеина из глутамата и цистеина, что является лимитирующей стадией синтеза GSH.
Впоследствии цистеин и глицин γ-глутаминовой кислоты катализируются GSH-синтазой (GSS) с образованием GSH (129).
GSH эффективно поддерживает Gpx в сокращенном состоянии.
У человека обнаружено восемь различных Gpx (Gpx1-8), а Gpx1-4 и Gpx6 представляют собой селенопротеины (131). По сравнению с другими членами семейства Gpx, Gpx4 представляет собой фермент, восстанавливающий перекисное окисление липидов, и он может превращать перекиси липидов (LOOH) в соответствующие спирты (LOH) (122, 132).
Следовательно, Gpx4 считается центральным ингибитором ферроптоза.
Многочисленные исследования показали, что ингибирование антиоксидантной системы Xc-GSH-Gpx4 может вызывать ферроптоз в клетках.
Например, эрастин, сульфасалазин (САВ) и сорафениб инициируют ферроптоз в клетках, ингибируя систему Xc (133, 134).
Бутионинсульфоксамин (BSO) индуцирует ферроптоз, ингибируя GCL (134), тогда как RSL3 может инициировать ферроптоз в клетках, ингибируя активность Gpx4 (135) (рис. 8).
Исследования последних лет показали, что ферроптоз тесно связан с гибелью эндотелиальных клеток. Например, Цинь и др. обнаружили, что наночастицы оксида цинка (ZnONP) могут индуцировать перекисное окисление железа и липидов в ЭК в зависимости от дозы и времени (136).
Далее команда применила липидный поглотитель активных форм кислорода ферростатин-1 и хелатор железа DFO для облегчения ZnONP-индуцированного ферроптоза в ЭК (22). Луо и др. показали, что ферроптоз связан с ЭД и что ось p53-xCT-GSH может регулировать процесс ферроптоза ЭК (137). Шэн и др. показали, что лизофосфатидилхолин (ЛФХ) может вызывать повышение внутриклеточного уровня железа и перекиси липидов, а также атрофию митохондрий при ЭК. Этот процесс можно обратить вспять астрагалозидом IV (AS-IV) (138).
Таким образом, ферроптоз является важным механизмом, с помощью которого АФК запускают запрограммированную гибель клеток при ЭК.