1. Введение
Сердечно-сосудистые заболевания (ССЗ) являются крупнейшим фактором глобальной смертности [1]. ССЗ охватывают множество заболеваний, включая атеросклероз, гипертонию, гиперактивность тромбоцитов, инсульт, гиперлипидемию и сердечную недостаточность [2]. Хотя генетические и другие состояния здоровья тесно связаны между собой, взаимодействие между диетой и микробиомом кишечника все чаще признается за их вклад в развитие и прогрессирование сердечно-сосудистых заболеваний. Несколько исследований показали связь между микробиотой кишечника и их метаболитами с факторами риска сердечно-сосудистых заболеваний, такими как гиперлипидемия, избыточный вес, воспаление, гипертония и гиперактивность тромбоцитов [3], подчеркивая сложную взаимосвязь между диетой, микробиотой кишечника и сердечно-сосудистыми заболеваниями [3,4]. Помимо своей роли в гемостазе и тромбозе, гиперактивные тромбоциты также являются важными медиаторами атеросклероза. Имеются убедительные доказательства гиперактивности тромбоцитов при таких состояниях, как диабет, курение, малоподвижный образ жизни, старение, ожирение, определенные метаболиты кишечника и нездоровое питание [5,6,7]. В организме человека обитают триллионы различных микробов, которые вместе называются микробиотой человека. Самая большая популяция микробов находится в кишечнике и содержит 100 триллионов микробов, по меньшей мере, 1000 различных видов бактерий. Достаточно данных указывает на то, что микробиом кишечника регулирует многочисленные физиологические функции, иммунную систему, сердечно-сосудистую систему, функцию кишечника, а также всасывание и метаболизм питательных веществ и их метаболитов. В нескольких исследованиях дисбактериоз кишечника связан с патологией сердечно-сосудистых заболеваний, включая атеросклероз, гипертонию, гиперактивность тромбоцитов, аномальный липидный обмен и сосудистую дисфункцию [8]. Дисбиоз кишечника является важным фактором, ответственным за критические факторы риска сердечно-сосудистых заболеваний, такие как атеросклероз, гипертония и гиперактивность тромбоцитов [9]. Появляющиеся данные свидетельствуют о том, что воздействие на микробиоту кишечника и ее метаболиты может быть эффективной стратегией лечения и профилактики сердечно-сосудистых заболеваний [9,10,11]. Различные виды кишечной микробиоты производят многочисленные метаболиты, в зависимости от рациона питания и состава микробиома, которые влияют на здоровье человека. Среди метаболитов кишечной микробиоты короткоцепочечные жирные кислоты (КЦЖК), вторичные метаболиты желчных кислот и триметил-N-оксид (ТМАО) являются важными модуляторными факторами различных заболеваний. Уровни ТМАО в плазме в значительной степени способствуют гиперактивности тромбоцитов, аномальным липидам плазмы, ожирению и резистентности к инсулину [3,8]. ТМАО увеличивает факторы риска сердечно-сосудистых заболеваний, изменяя метаболизм холестерина и желчных кислот, активируя воспалительные пути и способствуя образованию пенистых клеток и гиперактивации тромбоцитов, тогда как КЦЖК способствуют развитию атеросклероза и гипертонии по разным механизмам. Таким образом, важно исследовать клеточную передачу сигналов с участием метаболитов микробиоты кишечника в физиологических и патологических состояниях, чтобы понять их роль в здоровье и заболеваниях человека. Существует сложная связь между микробиотой кишечника, ее метаболитами и биологическими процессами, влияющими на риск сердечно-сосудистых заболеваний. Таким образом, углубленное понимание роли микробиоты кишечника и их метаболитов в прогрессировании и патогенезе сердечно-сосудистых заболеваний поможет нам развить терапевтический потенциал в будущем.
В этом обзоре описаны текущие данные, связывающие микробиоту кишечника и ее метаболиты с различными факторами риска сердечно-сосудистых заболеваний.
2. Роль кишечной микробиоты в процессе атеросклероза.
Процесс атеросклероза включает фиброз интимы, образование жировых бляшек, пролиферацию гладкомышечных клеток, миграцию моноцитов и Т-лимфоцитов, гиперактивность тромбоцитов и накопление холестерина [12]. Новые данные свидетельствуют о том, что дисбактериоз кишечника также может способствовать развитию атеросклероза за счет усиления системного воспаления [13,14,15]. Воспаление играет важную роль во многих заболеваниях, включая атеросклероз [12,16,17]. Накопленные данные указывают на то, что микробиота кишечника и их метаболиты играют важную роль в системном воспалении и модулируют различные факторы риска сердечно-сосудистых заболеваний [18]. Целостность кишечного барьера необходима для поддержания здоровья хозяина и предотвращения процессов воспаления и атеросклероза. Кишечная проницаемость нарушается из-за снижения экспрессии белков плотных соединений, таких как zonula occludens-1, claudin-1, окклюдин, и создания дисбаланса между гибелью и регенерацией эпителиальных клеток кишечника [19,20]. Akkermansia muciniphila оказывает защитное действие против атеросклероза, улучшая барьерные функции кишечника [21]. Анализ метагенома кишечника показал относительно более низкую численность Roseburia и Eubacterium, в то время как Collinsella была выше у пациентов с сердечно-сосудистыми заболеваниями, чем у здоровых людей [22]. Мета-анализ показал, что лечение антибиотиками не оказало существенного положительного эффекта при сердечно-сосудистых заболеваниях [23], хотя микробиота кишечника играет жизненно важную роль в воспалении и факторах риска сердечно-сосудистых заболеваний [18]. Целостность эпителия кишечника защищает от патогенной инвазии в большой круг кровообращения и, следовательно, от иммунных и воспалительных нарушений [24]. Когда целостность эпителия нарушена, вторжение патоген-ассоциированных молекулярных структур (PAMP) приводит к иммунному ответу и вызывает системное и тканеспецифическое воспаление. Некоторые PAMP могут стимулировать воспалительные процессы с участием рецепторов распознавания образов хозяина (PRR), таких как CpG-олигодезоксинуклеотиды, флагеллин и липопептиды [25]. Нарушение целостности кишечного барьера, вызванное дисбиозом кишечника, является значимым фактором риска хронического воспаления, наблюдаемого при ряде заболеваний, включая атеросклероз: микробный компонент, липополисахарид, является одним из PAMP, участвующих в развитии сердечно-сосудистых заболеваний. Связь между липополисахаридом и риском сердечно-сосудистых заболеваний впервые наблюдалась в 1999 году и определялась по уровням эндотоксинов у пациентов [26], а позже эта связь была подтверждена несколькими исследованиями [27,28]. Дисбиоз увеличивает проницаемость кишечника за счет подавления белков плотных соединений, что позволяет транслокировать липополисахарид в кровоток [29,30]. Липополисахарид, образующийся при дисбактериозе кишечника, связывает Toll-подобные рецепторы (TLR) и активирует последующие иммунные реакции [31]. Липополисахарид связывает комплекс TLR4 с его корецепторным кластером дифференцировки 14 (CD14). Повышение регуляции TLR инициирует воспалительный процесс атеросклероза [32,33]. Взаимодействие между липополисахаридом и TLR4 активирует пути MYD88 и NFκB, что приводит к усилению синтеза провоспалительных цитокинов, таких как IL-6, IL-1, IL-27 и TNF-α. Эти воспалительные цитокины участвуют в развитии атеросклероза и сердечно-сосудистых заболеваний [34,35]. Другой бактериальный PAMP, пептидогликан, увеличивает риск сердечно-сосудистых заболеваний, нарушая эпителиальный барьер кишечника. Метагеномное секвенирование показало, что у пациентов с атеросклерозом наблюдается обогащение генами, кодирующими синтез пептидогликана [22]. Наличие бактериального пептидогликана обнаружено в атеросклеротических бляшках [36]. Белки нуклеотидсвязывающего домена олигомеризации (NOD), NOD1 и NOD2, способствуют удалению внутриклеточных бактерий или бактериального мусора посредством распознавания пептидогликана с участием NFκB и MAP киназы [37]. NOD2 является важнейшим регулятором бактериального иммунитета и целостности барьера кишечника.
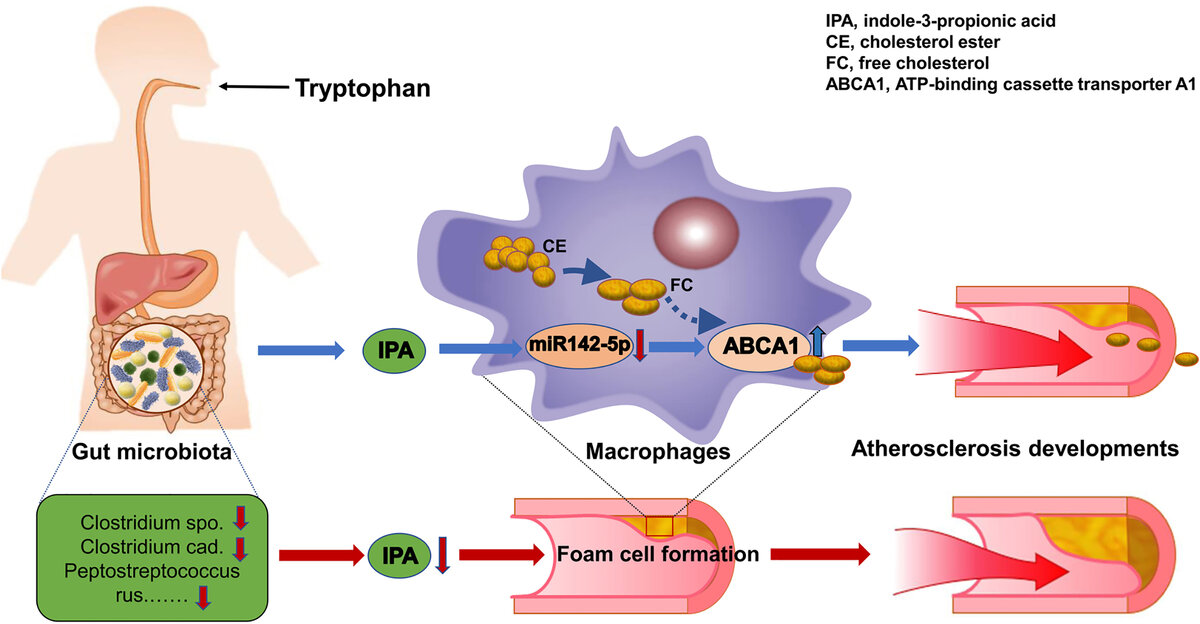
Изменения состава микрофлоры кишечника могут модулировать риск сердечно-сосудистых заболеваний. Несмотря на многочисленные данные, демонстрирующие вклад патогенных бактерий в развитие сердечно-сосудистых заболеваний, исследования антибиотиков дали неоднозначные результаты [38]. Метаанализ клинических исследований показал, что модификация микробиоты кишечника антибиотиками не продемонстрировала каких-либо преимуществ в отношении смертности от сердечно-сосудистых событий у пациентов с ишемической болезнью сердца [23]. Более того, в обширном исследовании с участием 4012 пациентов со стабильной ишемической болезнью сердца введение азитромицина не выявило влияния на риск сердечных событий [39]. Однако было показано, что состав микробиоты увеличивает тяжесть инфаркта миокарда на крысиной модели Dahl S с ишемией/реперфузионным повреждением сердца. Ванкомицин, плохо всасывающийся антибиотик, снижает частоту инфарктов миокарда на 27% и увеличивает постишемическое восстановление механических функций на 35% [40]. Этот эффект был связан с изменением микробиоты кишечника (как бактерий, так и грибов).
Это снижает уровень лептина в плазме, что позже это было подтверждено введением пробиотика, подавляющего лептин, Lactobacillus plantarum 299v [40]. Эти самые ранние противоречивые результаты относительно использования антибиотиков (азитромицин по сравнению с ванкомицином) объяснили сложность вмешательства на основе кишечной микробиоты с точки зрения эффективности и свойств применяемого протокола. Метаболиты кишечных микробов, такие как метиламины, полиамины, короткоцепочечные жирные кислоты (КЦЖК), триметиламин (ТМА) и вторичные желчные кислоты, играют важную роль в физиологии хозяина и в развитии сердечно-сосудистых заболеваний [41,42]. SCFAs, группа микробных продуктов (таких как пропионовая кислота, уксусная кислота и масляная кислота), играют решающую роль в возникновении и поддержании различных заболеваний [43]. Сообщалось о корреляции между повышенными уровнями ТМАО в плазме и атеросклерозом [44,45,46,47]. Участие этих микробных метаболитов в риске сердечно-сосудистых заболеваний на моделях как у людей, так и у животных было тщательно изучено [48]. В настоящее время имеется достаточно доказательств того, что кишечная микробиота участвует в здоровье кишечника, сердечно-сосудистой системы и иммунной функции.
Микробиом кишечника человека включает преимущественно пять типов: Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria и Verrucomicrobia [49].
В здоровом кишечнике анаэробные группы Bacteroidetes и Firmicutes составляют более 90% от общего числа видов бактерий [49]. Микробиом кишечника выполняет множество функций у хозяина [50]. Они участвуют в переваривании пищи человеком посредством двух основных катаболических сахаролитических и протеолитических катаболических путей. Оба пути приводят к производству SCFAs из полисахаридов. Второй катаболический путь также приводит к образованию аммиака, различных аминов, тиолов, фенолов и индолов. Микробиота кишечника метаболизирует пищевой фосфатидилхолин или L-карнитин в ТМА [41]. Затем ТМА попадает в печень и окисляется печеночной флавинмонооксигеназой 3 (FMO3), что приводит к образованию ТМАО [8]. Одним из них является ТМА, полученный из пищевых источников фосфатидилхолина, холина и L-карнитина. Синтез ТМАО является вторичным по отношению к приему пищевых компонентов, содержащих фрагмент ТМА, таких как холин, фосфатидилхолин и L-карнитин, которые в больших количествах присутствуют в продуктах животного происхождения, таких как красное мясо, рыба, молоко и яйца. Микробные лиазы ТМА метаболизируют эти соединения с образованием ТМА. Затем ТМА транспортируется в печень через портальную циркуляцию и окисляется в печени до ТМАО печеночными FMO, в первую очередь FMO3 [8,51]. ТМАО является уникальным среди традиционных факторов риска сердечно-сосудистых заболеваний, поскольку является продуктом метаболизма кишечных микробов. На уровень ТМАО в плазме влияют различные факторы, такие как диета, микрофлора кишечника, прием лекарств и активность флавинмонооксигеназы печени. ТМАО попадает в системный кровоток и способствует развитию атеросклероза, изменяя липидный обмен, активность тромбоцитов, ожирение и васкуляризацию (рис. 1).
ТМАО влияет на активность тромбоцитов, липидный обмен, ожирение, инсулинорезистентность, тонус сосудов и диабет, стимулируя тем самым процесс атеросклероза. Использование как пребиотиков, так и пробиотиков в комбинации или по отдельности влияет на состав микробиоты кишечника. Пребиотики, в том числе галактоолигосахариды, фруктоолигосахариды, инулин и др., стимулируют рост полезной микрофлоры, а пробиотики содержат специфические полезные штаммы бактерий. Эти вмешательства могут помочь модулировать бактерии для трансформации предшественников в ТМА и повышать способность бактерий истощать их, или могут помочь модулировать бактерии, лишенные генов, ответственных за преобразование карнитина или холина в ТМА. Большинство бактерий относятся к метанобактериям, обитающим в кишечнике человека. Было показано, что метаногенные бактерии истощают как ТМА, так и ТМАО [52,53]. Ресвератрол также может значительно модулировать рост специфической микробиоты кишечника in vivo, включая увеличение соотношения Bacteroidetes и Firmicutes и рост Bacteroides, Lactobacillus и Bifidobacterium [54,55]. Было показано, что эти изменения снижают уровень ТМАО. Снижение уровня L-карнитина или холина в рационе не является хорошей альтернативой, поскольку это важные питательные вещества.
ТМАО связано с ожирением, резистентностью к инсулину и заболеваниями почек [56]. Установлена связь между ТМАО и сердечно-сосудистыми заболеваниями [57]. Уровни ТМАО в крови хорошо коррелируют с размером атеросклеротических бляшек и сердечно-сосудистыми событиями [58].
Несколько метаанализов обнаружили связь уровней ТМАО в плазме с риском сердечно-сосудистых заболеваний и смертности [56,59]. Пациенты с самым высоким квартилем уровня циркулирующего ТМАО демонстрировали более высокий риск серьезных неблагоприятных сердечно-сосудистых событий, чем пациенты из нижнего квартиля [60]. Уровни ТМАО также были связаны с уязвимостью и образованием бляшек, долгосрочными рисками сердечно-сосудистых событий у пациентов и плохим прогнозом [61,62,63]. В исследованиях на животных диета с высоким содержанием холина вызывала повышение уровня ТМАО и атеросклероз. ТМАО опосредует, по крайней мере частично, установленную связь между потреблением красного мяса и риском сердечно-сосудистых заболеваний. Следовательно, низкий уровень ТМАО в крови в результате потребления фруктов и овощей может объяснять их кардиопротекторное действие. Продолжается работа по поиску новых терапевтических подходов, снижающих уровень ТМАО в плазме. Имеющиеся данные указывают на модуляцию пути генерации ТМАО микробами кишечника, который ослабляет атеросклероз и гиперактивность тромбоцитов, а также потенциал тромбоза in vivo на животных моделях; однако эффективный метод лечения еще не установлен. Модификация образа жизни, включая физические упражнения, диету, функциональное питание и изменение микробиоты, может быть полезна для снижения уровня ТМАО [64]. ТМАО усугубляет воспалительные реакции сосудистой стенки, индуцирует выработку АФК и предотвращает обратный транспорт холестерина [65]. Модулированный ТМАО метаболизм холестерина и стеринов способствует развитию атеросклероза [46]. У мышей с нокдауном FMO3 снижались уровни циркулирующего ТМАО и ослаблялось образование атеросклерозных бляшек, несмотря на активацию обратного транспорта холестерина макрофагами [66,67,68]. Уровни в плазме кишечных микробных пищевых метаболитов фосфатидилхолина и ТМАО, продуцирующих l-карнитин и γ-бутиробетаин, были связаны с риском сердечно-сосудистых заболеваний [69,70,71]. Уровень ТМАО в плазме был напрямую связан с образованием атеросклеротических бляшек [8]. Проспективное наблюдательное клиническое исследование пациентов с хронической сердечной недостаточностью или без нее последовательно показало, что уровни ТМАО в плазме положительно коррелируют с риском сердечной недостаточности [72]. Роль ТМАО в развитии атеросклероза исследовали с использованием пищевой добавки холина на мышах ApoE-/-. Были измерены CD36, стероидный рецептор РНК-активатора 1 (SR-A1) и 2 рецептора-поглотителя макрофагов. Уровни CD36 и SR-A1 в макрофагах мышей, получавших ТМАО, были повышены по сравнению с контрольной группой, а введение антибиотиков уменьшало образование пенистых клеток за счет снижения продукции ТМА [8]. Существенного влияния ТМАО на образование пенистых ячеек не наблюдалось. ТМАО также способствует развитию атеросклероза, подавляя обратный транспорт холестерина и изменяя активность переносчиков холестерина в макрофагах [69]. Кроме того, ТМАО подавляет экспрессию синтетазы желчных кислот Cyp7a1 и Cyp27a1 печени и переносчиков желчных кислот Oatp1, Oatp4, Mrp2 и Ntcp, что приводит к нарушению путей, связанных с желчными кислотами, и способствует атеросклерозу [71]. Рецептор фарнезоида X (FXR) также контролирует метаболизм желчных кислот и выработку ТМАО, регулируя экспрессию печеночного FMO3 [51]. FXR защищал мышей от атеросклероза путем ингибирования экспрессии CYP7A1 и CYP8B1 у мышей ApoE-/- [51,66,73,74].
ТМАО ускоряет атеросклероз за счет нескольких механизмов, таких как усиление притока холестерина, ингибирование оттока холестерина, блокирование пути желчных кислот и вызывание гиперактивности тромбоцитов. ТМАО также повышал экспрессию молекулы адгезии сосудистых клеток-1 (VCAM-1) и активировал протеинкиназа C (PKC) и NFκB. Таким образом, ТМАО стимулирует атеросклероз посредством дисфункции эндотелиальных клеток и увеличивает адгезию моноцитов [75]. Эти результаты подтвердили роль ТМАО в развитии сердечно-сосудистых заболеваний. Однако необходима дальнейшая работа, чтобы установить, как ТМАО можно использовать в качестве биомаркера риска сердечно-сосудистых заболеваний и атеросклеротических заболеваний [76,77,78]. ТМАО можно рассматривать как независимый фактор риска сердечно-сосудистых заболеваний. Однако также наблюдались противоречивые результаты, особенно при широких популяционных наблюдениях [79,80].
Холин обычно считается пищевым источником ТМАО; однако не было обнаружено существенных доказательств значимой связи между потреблением холина и риском сердечно-сосудистых заболеваний [81]. Введение l-карнитина приводило к значительному увеличению уровня циркулирующего ТМАО у мышей ApoE(-/-), но его статус обратно коррелировал с размером поражения аорты [82]. Несколько популяционных исследований в разных странах показали, что потребление холина и бетаина с пищей не связано с патогенезом сердечно-сосудистых заболеваний [79,83]. Однако для окончательных выводов о точной роли ТМАО при атеросклерозе, а также подтверждения его терапевтического потенциала путем воздействия на бактерии или ферменты, продуцирующие ТМАО, необходимы более глубокие исследования. Другой метаболит, полученный из кишечной микробиоты, желчная кислота, участвует в различных метаболических заболеваниях [84]. Желчная кислота накапливается в желчном пузыре и высвобождается в кишечник, помогая всасыванию пищевых липидов и жирорастворимых витаминов. Первичные желчные кислоты обычно метаболизируются ферментами кишечной микробиоты во вторичные желчные кислоты, в том числе дезоксихолевая кислота, литохолевая кислота, гиодеоксихолевая кислота и урсодезоксихолевая кислота [85].
Подавление биосинтеза желчных кислот в печени может также ингибировать изменения микробиома кишечника, вызванные диетой с высоким содержанием жиров, что указывает на наличие метаболической оси печень-желчные кислоты-кишечник микробиом [86]. Недавно была рассмотрена двунаправленная связь между микробиотой кишечника и метаболизмом желчных кислот [87] при сердечно-сосудистых заболеваниях [48]. Желчные кислоты могут ускорять развитие атеросклероза за счет активности гидролазы желчных солей и рецепторов желчных кислот [88,89]. Гидролаза желчных солей присутствует у многих бактерий и архей, таких как Methanobrevibacter smithii, Clostridium, Enterococcus [90,91]. Бактериально-опосредованная активность солевой гидролазы желчных кислот может способствовать прогрессированию атеросклеротического процесса, стимулируя накопление холестерина, образование пенистых клеток и увеличение размера атеросклеротических бляшек [92]. TGR5, рецептор, связанный с G-белком, является важным рецептором желчных кислот хозяина, который опосредует системные эффекты желчных кислот [93]. TGR5 может ингибировать развитие атеросклероза, уменьшая опосредованное макрофагами воспаление и липидную нагрузку [94]. Рецептор прегнана X (PXR) также регулирует экспрессию генов, участвующих в биосинтезе, транспорте и метаболизме желчных кислот. PXR активируется вторичными желчными кислотами [95]. Активация PXR увеличивает атерогенные липопротеины ЛПОНП и ЛПНП [96]. Развитие атеросклероза задерживается у мышей с двойным нокаутом PXR и apoE (PXR-/- и ApoE-/-) [97]. Вторичные желчные кислоты, полученные из микробиоты, играют важную роль в развитии атеросклероза, модулируя различные рецепторы желчных кислот, такие как FXR, PXR, TGR5 и VDR, а также S1PR2. Благоприятная модуляция метаболизма желчных кислот путем воздействия на микробиоту также может предотвратить процесс атеросклероза [98].
Выводы
Новые данные свидетельствуют о тесной связи между соединениями, полученными из микробиоты, и повышенным риском сердечно-сосудистых заболеваний. Поэтому важно продолжить изучение роли диет, микробного производства ТМАО и SCFAs, а также их клеточной передачи сигналов, чтобы определить их влияние на сердечно-сосудистую физиологию. В настоящее время установлена связь между гиперактивностью тромбоцитов и сердечно-сосудистыми заболеваниями. Учитывая растущую озабоченность сердечно-сосудистыми заболеваниями из-за гиперактивности тромбоцитов, наблюдаемой при ожирении, нарушении липидного обмена, малоподвижном образе жизни, резистентности к инсулину, ТМАО и многих других, новая терапия пробиотиками и пребиотиками может представлять собой подходящий режим первичной профилактики без чрезмерного снижения питательно важных прием предшественников ТМА, таких как холин, бетаин и L-карнитин.
Хотя для лечения сердечно-сосудистых заболеваний существует несколько препаратов, в настоящее время они являются основной причиной смертности во всем мире. Появилось достаточно убедительных данных о связи дисбактериоза кишечника и сердечно-сосудистых заболеваний. Тем не менее, необходима дальнейшая работа для создания таргетной терапии сердечно-сосудистых заболеваний, направленной на микробиоту кишечника. Поскольку доступны различные экспериментальные и клинические данные о механизмах развития сердечно-сосудистых заболеваний, опосредованных микробиотой кишечника, существует большая вероятность найти новые подходы к лечению или профилактике сердечно-сосудистых заболеваний. SCFAs и некоторые типы желчных кислот или снижение уровня микробного метаболита ТМА можно модулировать с помощью диеты, пребиотиков и пробиотиков, а также с помощью специфических ингибиторов ТМА.
Микробный состав кишечника также можно благоприятно модифицировать с помощью пробиотиков, пребиотиков и натуральных компонентов. С этой целью необходимы хорошо спланированные крупномасштабные клинические исследования для подтверждения данных доклинических и других небольших исследований на людях. Доступные в настоящее время данные позволяют нам воздействовать на микробиоту кишечника и ее метаболиты, чтобы понять механизмы сердечно-сосудистых заболеваний и разработать новые профилактические или терапевтические режимы.
1. Kwan G.F., Mayosi B.M., Mocumbi A.O., Miranda J.J., Ezzati M., Jain Y., Robles G., Benjamin E.J., Subramanian S.V., Bukhman G. Endemic Cardiovascular Diseases of the Poorest Billion. Circulation. 2016;133:2561–2575. doi: 10.1161/CIRCULATIONAHA.116.008731. [PubMed] [CrossRef] [Google Scholar]
2. Writing Group M., Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., Das S.R., de Ferranti S., Despres J.P., et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [PubMed] [CrossRef] [Google Scholar]
3. Zhu W., Gregory J.C., Org E., Buffa J.A., Gupta N., Wang Z., Li L., Fu X., Wu Y., Mehrabian M., et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
4. Wang H., Zhang W., Zuo L., Dong J., Zhu W., Li Y., Gu L., Gong J., Li Q., Li N., et al. Intestinal dysbacteriosis contributes to decreased intestinal mucosal barrier function and increased bacterial translocation. Lett. Appl. Microbiol. 2014;58:384–392. doi: 10.1111/lam.12201. [PubMed] [CrossRef] [Google Scholar]
5. Willoughby S., Holmes A., Loscalzo J. Platelets and cardiovascular disease. Eur. J. Cardiovasc. Nurs. 2002;1:273–288. doi: 10.1016/S1474-5151(02)00038-5. [PubMed] [CrossRef] [Google Scholar]
6. Michelson A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010;9:154–169. doi: 10.1038/nrd2957. [PubMed] [CrossRef] [Google Scholar]
7. Jackson S.P., Nesbitt W.S., Westein E. Dynamics of platelet thrombus formation. J. Thromb. Haemost. 2009;7(Suppl. 1):17–20. doi: 10.1111/j.1538-7836.2009.03401.x. [PubMed] [CrossRef] [Google Scholar]
8. Wang Z., Klipfell E., Bennett B.J., Koeth R., Levison B.S., Dugar B., Feldstein A.E., Britt E.B., Fu X., Chung Y.M., et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Lau K., Srivatsav V., Rizwan A., Nashed A., Liu R., Shen R., Akhtar M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients. 2017;9:859. doi: 10.3390/nu9080859. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
10. Anbazhagan A.N., Priyamvada S., Priyadarshini M. Gut Microbiota in Vascular Disease: Therapeutic Target? Curr. Vasc. Pharm. 2017;15:291–295. doi: 10.2174/1570161115666170105095834. [PubMed] [CrossRef] [Google Scholar]
11. Santisteban M.M., Qi Y., Zubcevic J., Kim S., Yang T., Shenoy V., Cole-Jeffrey C.T., Lobaton G.O., Stewart D.C., Rubiano A., et al. Hypertension-Linked Pathophysiological Alterations in the Gut. Circ. Res. 2017;120:312–323. doi: 10.1161/CIRCRESAHA.116.309006. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
12. Gui T., Shimokado A., Sun Y., Akasaka T., Muragaki Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to biomarker discovery. Mediat. Inflamm. 2012;2012:693083. doi: 10.1155/2012/693083. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
13. Drosos I., Tavridou A., Kolios G. New aspects on the metabolic role of intestinal microbiota in the development of atherosclerosis. Metabolism. 2015;64:476–481. doi: 10.1016/j.metabol.2015.01.007. [PubMed] [CrossRef] [Google Scholar]
14. Gregory J.C., Buffa J.A., Org E., Wang Z., Levison B.S., Zhu W., Wagner M.A., Bennett B.J., Li L., DiDonato J.A., et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
15. Jie Z., Xia H., Zhong S.L., Feng Q., Li S., Liang S., Zhong H., Liu Z., Gao Y., Zhao H., et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017;8:845. doi: 10.1038/s41467-017-00900-1. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
16. Xu H., Barnes G.T., Yang Q., Tan G., Yang D., Chou C.J., Sole J., Nichols A., Ross J.S., Tartaglia L.A., et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003;112:1821–1830. doi: 10.1172/JCI200319451. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
17. Ding S., Chi M.M., Scull B.P., Rigby R., Schwerbrock N.M., Magness S., Jobin C., Lund P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
18. Kasahara K., Tanoue T., Yamashita T., Yodoi K., Matsumoto T., Emoto T., Mizoguchi T., Hayashi T., Kitano N., Sasaki N., et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J. Lipid Res. 2017;58:519–528. doi: 10.1194/jlr.M072165. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
19. Wang Z., Tang W.H., Buffa J.A., Fu X., Britt E.B., Koeth R.A., Levison B.S., Fan Y., Wu Y., Hazen S.L. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur. Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Chen W.Y., Wang M., Zhang J., Barve S.S., McClain C.J., Joshi-Barve S. Acrolein Disrupts Tight Junction Proteins and Causes Endoplasmic Reticulum Stress-Mediated Epithelial Cell Death Leading to Intestinal Barrier Dysfunction and Permeability. Am. J. Pathol. 2017;187:2686–2697. doi: 10.1016/j.ajpath.2017.08.015. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
21. Li J., Lin S., Vanhoutte P.M., Woo C.W., Xu A. Akkermansia Muciniphila Protects Against Atherosclerosis by Preventing Metabolic Endotoxemia-Induced Inflammation in Apoe−/− Mice. Circulation. 2016;133:2434–2446. doi: 10.1161/CIRCULATIONAHA.115.019645. [PubMed] [CrossRef] [Google Scholar]
22. Karlsson F.H., Fak F., Nookaew I., Tremaroli V., Fagerberg B., Petranovic D., Backhed F., Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
23. Andraws R., Berger J.S., Brown D.L. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: A meta-analysis of randomized controlled trials. JAMA. 2005;293:2641–2647. doi: 10.1001/jama.293.21.2641. [PubMed] [CrossRef] [Google Scholar]
24. Desai M.S., Seekatz A.M., Koropatkin N.M., Kamada N., Hickey C.A., Wolter M., Pudlo N.A., Kitamoto S., Terrapon N., Muller A., et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell. 2016;167:1339–1353.e1321. doi: 10.1016/j.cell.2016.10.043. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
25. Kholy K.E., Genco R.J., Van Dyke T.E. Oral infections and cardiovascular disease. Trends Endocrinol. Metab. 2015;26:315–321. doi: 10.1016/j.tem.2015.03.001. [PubMed] [CrossRef] [Google Scholar]
26. Wiedermann C.J., Kiechl S., Dunzendorfer S., Schratzberger P., Egger G., Oberhollenzer F., Willeit J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 1999;34:1975–1981. doi: 10.1016/S0735-1097(99)00448-9. [PubMed] [CrossRef] [Google Scholar]
27. Miller M.A., McTernan P.G., Harte A.L., Silva N.F., Strazzullo P., Alberti K.G., Kumar S., Cappuccio F.P. Ethnic and sex differences in circulating endotoxin levels: A novel marker of atherosclerotic and cardiovascular risk in a British multi-ethnic population. Atherosclerosis. 2009;203:494–502. doi: 10.1016/j.atherosclerosis.2008.06.018. [PubMed] [CrossRef] [Google Scholar]
28. Mitra S., Drautz-Moses D.I., Alhede M., Maw M.T., Liu Y., Purbojati R.W., Yap Z.H., Kushwaha K.K., Gheorghe A.G., Bjarnsholt T., et al. In silico analyses of metagenomes from human atherosclerotic plaque samples. Microbiome. 2015;3:38. doi: 10.1186/s40168-015-0100-y. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
29. Cani P.D., Bibiloni R., Knauf C., Waget A., Neyrinck A.M., Delzenne N.M., Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [PubMed] [CrossRef] [Google Scholar]
30. Harris K., Kassis A., Major G., Chou C.J. Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J. Obes. 2012;2012:879151. doi: 10.1155/2012/879151. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Chacon M.R., Lozano-Bartolome J., Portero-Otin M., Rodriguez M.M., Xifra G., Puig J., Blasco G., Ricart W., Chaves F.J., Fernandez-Real J.M. The gut mycobiome composition is linked to carotid atherosclerosis. Benef. Microbes. 2018;9:185–198. doi: 10.3920/BM2017.0029. [PubMed] [CrossRef] [Google Scholar]
32. Xu X.H., Shah P.K., Faure E., Equils O., Thomas L., Fishbein M.C., Luthringer D., Xu X.P., Rajavashisth T.B., Yano J., et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [PubMed] [CrossRef] [Google Scholar]
33. Edfeldt K., Swedenborg J., Hansson G.K., Yan Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation. 2002;105:1158–1161. doi: 10.1161/circ.105.10.1158. [PubMed] [CrossRef] [Google Scholar]
34. Barton G.M., Kagan J.C. A cell biological view of Toll-like receptor function: Regulation through compartmentalization. Nat. Rev. Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
35. Guzzo C., Ayer A., Basta S., Banfield B.W., Gee K. IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 2012;188:864–873. doi: 10.4049/jimmunol.1101912. [PubMed] [CrossRef] [Google Scholar]
36. Laman J.D., Schoneveld A.H., Moll F.L., van Meurs M., Pasterkamp G. Significance of peptidoglycan, a proinflammatory bacterial antigen in atherosclerotic arteries and its association with vulnerable plaques. Am. J. Cardiol. 2002;90:119–123. doi: 10.1016/S0002-9149(02)02432-3. [PubMed] [CrossRef] [Google Scholar]
37. Philpott D.J., Sorbara M.T., Robertson S.J., Croitoru K., Girardin S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [PubMed] [CrossRef] [Google Scholar]
38. Munford R.S. Endotoxemia-menace, marker, or mistake? J. Leukoc. Biol. 2016;100:687–698. doi: 10.1189/jlb.3RU0316-151R. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
39. Grayston J.T., Kronmal R.A., Jackson L.A., Parisi A.F., Muhlestein J.B., Cohen J.D., Rogers W.J., Crouse J.R., Borrowdale S.L., Schron E., et al. Azithromycin for the secondary prevention of coronary events. N. Engl. J. Med. 2005;352:1637–1645. doi: 10.1056/NEJMoa043526. [PubMed] [CrossRef] [Google Scholar]
40. Lam V., Su J., Koprowski S., Hsu A., Tweddell J.S., Rafiee P., Gross G.J., Salzman N.H., Baker J.E. Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J. 2012;26:1727–1735. doi: 10.1096/fj.11-197921. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
41. Brown J.M., Hazen S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015;66:343–359. doi: 10.1146/annurev-med-060513-093205. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
42. Bergeron N., Williams P.T., Lamendella R., Faghihnia N., Grube A., Li X., Wang Z., Knight R., Jansson J.K., Hazen S.L., et al. Diets high in resistant starch increase plasma levels of trimethylamine-N-oxide, a gut microbiome metabolite associated with CVD risk. Br. J. Nutr. 2016;116:2020–2029. doi: 10.1017/S0007114516004165. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
43. Li X., Shimizu Y., Kimura I. Gut microbial metabolite short-chain fatty acids and obesity. Biosci. Microbiota Food Health. 2017;36:135–140. doi: 10.12938/bmfh.17-010. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
44. Randrianarisoa E., Lehn-Stefan A., Wang X., Hoene M., Peter A., Heinzmann S.S., Zhao X., Konigsrainer I., Konigsrainer A., Balletshofer B., et al. Relationship of Serum Trimethylamine N-Oxide (TMAO) Levels with early Atherosclerosis in Humans. Sci. Rep. 2016;6:26745. doi: 10.1038/srep26745. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
45. Wang Z., Roberts A.B., Buffa J.A., Levison B.S., Zhu W., Org E., Gu X., Huang Y., Zamanian-Daryoush M., Culley M.K., et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163:1585–1595. doi: 10.1016/j.cell.2015.11.055. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
46. Koeth R.A., Wang Z., Levison B.S., Buffa J.A., Org E., Sheehy B.T., Britt E.B., Fu X., Wu Y., Li L., et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013;19:576–585. doi: 10.1038/nm.3145. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
47. Li X.S., Obeid S., Klingenberg R., Gencer B., Mach F., Raber L., Windecker S., Rodondi N., Nanchen D., Muller O., et al. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: A prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur. Heart J. 2017;38:814–824. doi: 10.1093/eurheartj/ehw582. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
48. Brown J.M., Hazen S.L. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018;16:171–181. doi: 10.1038/nrmicro.2017.149. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
49. Qin J., Li R., Raes J., Arumugam M., Burgdorf K.S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
50. Backhed F., Ley R.E., Sonnenburg J.L., Peterson D.A., Gordon J.I. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [PubMed] [CrossRef] [Google Scholar]
51. Bennett B.J., de Aguiar Vallim T.Q., Wang Z., Shih D.M., Meng Y., Gregory J., Allayee H., Lee R., Graham M., Crooke R., et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
52. Brugere J.F., Borrel G., Gaci N., Tottey W., O’Toole P.W., Malpuech-Brugere C. Archaebiotics: Proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes. 2014;5:5–10. doi: 10.4161/gmic.26749. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
53. Dridi B., Fardeau M.L., Ollivier B., Raoult D., Drancourt M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2012;62:1902–1907. doi: 10.1099/ijs.0.033712-0. [PubMed] [CrossRef] [Google Scholar]
54. Chen M.L., Yi L., Zhang Y., Zhou X., Ran L., Yang J., Zhu J.D., Zhang Q.Y., Mi M.T. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. MBio. 2016;7:e02210–e02215. doi: 10.1128/mBio.02210-15. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
55. Larrosa M., Yanez-Gascon M.J., Selma M.V., Gonzalez-Sarrias A., Toti S., Ceron J.J., Tomas-Barberan F., Dolara P., Espin J.C. Effect of a low dose of dietary resveratrol on colon microbiota, inflammation and tissue damage in a DSS-induced colitis rat model. J. Agric. Food Chem. 2009;57:2211–2220. doi: 10.1021/jf803638d. [PubMed] [CrossRef] [Google Scholar]
56. Sun G., Yin Z., Liu N., Bian X., Yu R., Su X., Zhang B., Wang Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017;493:964–970. doi: 10.1016/j.bbrc.2017.09.108. [PubMed] [CrossRef] [Google Scholar]
57. Bu J., Wang Z. Cross-Talk between Gut Microbiota and Heart via the Routes of Metabolite and Immunity. Gastroenterol. Res. Pr. 2018;2018:6458094. doi: 10.1155/2018/6458094. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
58. Qi J., You T., Li J., Pan T., Xiang L., Han Y., Zhu L. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: A systematic review and meta-analysis of 11 prospective cohort studies. J. Cell. Mol. Med. 2018;22:185–194. doi: 10.1111/jcmm.13307. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
59. Heianza Y., Ma W., Manson J.E., Rexrode K.M., Qi L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Heart Assoc. 2017;6:e004947. doi: 10.1161/JAHA.116.004947. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
60. Tang W.H., Wang Z., Levison B.S., Koeth R.A., Britt E.B., Fu X., Wu Y., Hazen S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
61. Fu Q., Zhao M., Wang D., Hu H., Guo C., Chen W., Li Q., Zheng L., Chen B. Coronary Plaque Characterization Assessed by Optical Coherence Tomography and Plasma Trimethylamine-N-oxide Levels in Patients With Coronary Artery Disease. Am. J. Cardiol. 2016;118:1311–1315. doi: 10.1016/j.amjcard.2016.07.071. [PubMed] [CrossRef] [Google Scholar]
62. Zhu W., Buffa J.A., Wang Z., Warrier M., Schugar R., Shih D.M., Gupta N., Gregory J.C., Org E., Fu X., et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J. Thromb. Haemost. 2018;16:1857–1872. doi: 10.1111/jth.14234. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
63. Suzuki T., Heaney L.M., Jones D.J., Ng L.L. Trimethylamine N-oxide and Risk Stratification after Acute Myocardial Infarction. Clin. Chem. 2017;63:420–428. doi: 10.1373/clinchem.2016.264853. [PubMed] [CrossRef] [Google Scholar]
64. Janeiro M.H., Ramirez M.J., Milagro F.I., Martinez J.A., Solas M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients. 2018;10:1398. doi: 10.3390/nu10101398. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
65. Sun X., Jiao X., Ma Y., Liu Y., Zhang L., He Y., Chen Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016;481:63–70. doi: 10.1016/j.bbrc.2016.11.017. [PubMed] [CrossRef] [Google Scholar]
66. Miao J., Morbid Obesity Study Group. Ling A.V., Manthena P.V., Gearing M.E., Graham M.J., Crooke R.M., Croce K.J., Esquejo R.M., Clish C.B., et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015;6:6498. doi: 10.1038/ncomms7498. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
67. Shih D.M., Wang Z., Lee R., Meng Y., Che N., Charugundla S., Qi H., Wu J., Pan C., Brown J.M., et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 2015;56:22–37. doi: 10.1194/jlr.M051680. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
68. Warrier M., Shih D.M., Burrows A.C., Ferguson D., Gromovsky A.D., Brown A.L., Marshall S., McDaniel A., Schugar R.C., Wang Z., et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015;10:326–338. doi: 10.1016/j.celrep.2014.12.036. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
69. Koeth R.A., Levison B.S., Culley M.K., Buffa J.A., Wang Z., Gregory J.C., Org E., Wu Y., Li L., Smith J.D., et al. gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014;20:799–812. doi: 10.1016/j.cmet.2014.10.006. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
70. Chen K., Zheng X., Feng M., Li D., Zhang H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017;8:139. doi: 10.3389/fphys.2017.00139. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
71. Guasch-Ferre M., Hu F.B., Ruiz-Canela M., Bullo M., Toledo E., Wang D.D., Corella D., Gomez-Gracia E., Fiol M., Estruch R., et al. Plasma Metabolites From Choline Pathway and Risk of Cardiovascular Disease in the PREDIMED (Prevention With Mediterranean Diet) Study. J. Am. Heart Assoc. 2017;6:e006524. doi: 10.1161/JAHA.117.006524. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
72. Troseid M., Ueland T., Hov J.R., Svardal A., Gregersen I., Dahl C.P., Aakhus S., Gude E., Bjorndal B., Halvorsen B., et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J. Intern. Med. 2015;277:717–726. doi: 10.1111/joim.12328. [PubMed] [CrossRef] [Google Scholar]
73. Mencarelli A., Renga B., Distrutti E., Fiorucci S. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Heart Circ. Physiol. 2009;296:H272–H281. doi: 10.1152/ajpheart.01075.2008. [PubMed] [CrossRef] [Google Scholar]
74. Miyazaki-Anzai S., Masuda M., Levi M., Keenan A.L., Miyazaki M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS ONE. 2014;9:e108270. doi: 10.1371/journal.pone.0108270. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
75. Ma G., Pan B., Chen Y., Guo C., Zhao M., Zheng L., Chen B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017;37:BSR20160244. doi: 10.1042/BSR20160244. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
76. Senthong V., Li X.S., Hudec T., Coughlin J., Wu Y., Levison B., Wang Z., Hazen S.L., Tang W.H. Plasma Trimethylamine N-Oxide, a Gut Microbe-Generated Phosphatidylcholine Metabolite, Is Associated With Atherosclerotic Burden. J. Am. Coll. Cardiol. 2016;67:2620–2628. doi: 10.1016/j.jacc.2016.03.546. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
77. Senthong V., Wang Z., Li X.S., Fan Y., Wu Y., Tang W.H., Hazen S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Heart Assoc. 2016;5:e002816. doi: 10.1161/JAHA.115.002816. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
78. Zheng Y., Li Y., Rimm E.B., Hu F.B., Albert C.M., Rexrode K.M., Manson J.E., Qi L. Dietary phosphatidylcholine and risk of all-cause and cardiovascular-specific mortality among US women and men. Am. J. Clin. Nutr. 2016;104:173–180. doi: 10.3945/ajcn.116.131771. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
79. Dalmeijer G.W., Olthof M.R., Verhoef P., Bots M.L., van der Schouw Y.T. Prospective study on dietary intakes of folate, betaine, and choline and cardiovascular disease risk in women. Eur. J. Clin. Nutr. 2008;62:386–394. doi: 10.1038/sj.ejcn.1602725. [PubMed] [CrossRef] [Google Scholar]
80. Meyer K.A., Benton T.Z., Bennett B.J., Jacobs D.R., Jr., Lloyd-Jones D.M., Gross M.D., Carr J.J., Gordon-Larsen P., Zeisel S.H. Microbiota-Dependent Metabolite Trimethylamine N-Oxide and Coronary Artery Calcium in the Coronary Artery Risk Development in Young Adults Study (CARDIA) J. Am. Heart Assoc. 2016;5:e003970. doi: 10.1161/JAHA.116.003970. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
81. Nagata C., Wada K., Tamura T., Konishi K., Kawachi T., Tsuji M., Nakamura K. Choline and Betaine Intakes Are Not Associated with Cardiovascular Disease Mortality Risk in Japanese Men and Women. J. Nutr. 2015;145:1787–1792. doi: 10.3945/jn.114.209296. [PubMed] [CrossRef] [Google Scholar]
82. Collins H.L., Drazul-Schrader D., Sulpizio A.C., Koster P.D., Williamson Y., Adelman S.J., Owen K., Sanli T., Bellamine A. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP. Atherosclerosis. 2016;244:29–37. doi: 10.1016/j.atherosclerosis.2015.10.108. [PubMed] [CrossRef] [Google Scholar]
83. Bidulescu A., Chambless L.E., Siega-Riz A.M., Zeisel S.H., Heiss G. Usual choline and betaine dietary intake and incident coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc. Disord. 2007;7:20. doi: 10.1186/1471-2261-7-20. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
84. Parseus A., Sommer N., Sommer F., Caesar R., Molinaro A., Stahlman M., Greiner T.U., Perkins R., Backhed F. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66:429–437. doi: 10.1136/gutjnl-2015-310283. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
85. Russell D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [PubMed] [CrossRef] [Google Scholar]
86. Zheng X., Huang F., Zhao A., Lei S., Zhang Y., Xie G., Chen T., Qu C., Rajani C., Dong B., et al. Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice. BMC Biol. 2017;15:120. doi: 10.1186/s12915-017-0462-7. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
87. Jones M.L., Martoni C.J., Ganopolsky J.G., Labbe A., Prakash S. The human microbiome and bile acid metabolism: Dysbiosis, dysmetabolism, disease and intervention. Expert Opin. Biol. Ther. 2014;14:467–482. doi: 10.1517/14712598.2014.880420. [PubMed] [CrossRef] [Google Scholar]
88. Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [PubMed] [CrossRef] [Google Scholar]
89. Ridlon J.M., Harris S.C., Bhowmik S., Kang D.J., Hylemon P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7:22–39. doi: 10.1080/19490976.2015.1127483. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
90. Jones M.L., Martoni C.J., Parent M., Prakash S. Cholesterol-lowering efficacy of a microencapsulated bile salt hydrolase-active Lactobacillus reuteri NCIMB 30242 yoghurt formulation in hypercholesterolaemic adults. Br. J. Nutr. 2012;107:1505–1513. doi: 10.1017/S0007114511004703. [PubMed] [CrossRef] [Google Scholar]
91. Tremaroli V., Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [PubMed] [CrossRef] [Google Scholar]
92. Hansson G.K., Robertson A.K., Soderberg-Naucler C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [PubMed] [CrossRef] [Google Scholar]
93. Li T., Chiang J.Y. Bile acids as metabolic regulators. Curr. Opin. Gastroenterol. 2015;31:159–165. doi: 10.1097/MOG.0000000000000156. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
94. Pols T.W., Nomura M., Harach T., Lo Sasso G., Oosterveer M.H., Thomas C., Rizzo G., Gioiello A., Adorini L., Pellicciari R., et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
95. Staudinger J.L., Goodwin B., Jones S.A., Hawkins-Brown D., MacKenzie K.I., LaTour A., Liu Y., Klaassen C.D., Brown K.K., Reinhard J., et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
96. Zhou C., King N., Chen K.Y., Breslow J.L. Activation of PXR induces hypercholesterolemia in wild-type and accelerates atherosclerosis in apoE deficient mice. J. Lipid Res. 2009;50:2004–2013. doi: 10.1194/jlr.M800608-JLR200. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
97. Sui Y., Xu J., Rios-Pilier J., Zhou C. Deficiency of PXR decreases atherosclerosis in apoE-deficient mice. J. Lipid Res. 2011;52:1652–1659. doi: 10.1194/jlr.M017376. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
98. Levi M. Role of Bile Acid-Regulated Nuclear Receptor FXR and G Protein-Coupled Receptor TGR5 in Regulation of Cardiorenal Syndrome (Cardiovascular Disease and Chronic Kidney Disease) Hypertension. 2016;67:1080–1084. doi: 10.1161/HYPERTENSIONAHA.115.06417. [PMC free article] [PubMed] [CrossRef] [Google Scholar]