Машинный перевод и адаптация интересной статьи зарубежного издания на тему похудения и длительного голодания у полных людей, страдающих ожирением. Не является рекомендацией!

Замечательная способность организма адаптироваться к длительному голоданию имела решающее значение для выживания первобытного человека. Понимание этих процессов может дать клиницисту лучшее понимание многих клинических состояний, характеризующихся кетоацидозом.
Краткое содержание: Организм адаптируется к длительному голоданию, сохраняя азот, поскольку мозг все чаще использует кетокислоты, избавляясь от потребности в глюкозе. Этот сдвиг в использовании топлива снижает потребность в мобилизации аминокислот из мышц для целей глюконеогенеза. Потеря азота с мочой первоначально происходит в виде мочевины, когда доминирует печеночный глюконеогенез, а затем в виде аммиака, что отражает повышенное поглощение глютамина почками. Углеродный скелет глутамина используется для производства глюкозы и регенерации израсходованного HCO 3 - . Замена мочевины на NH 4+обеспечивает осмоли, необходимые для оттока мочи и выведения продуктов жизнедеятельности. Со временем потеря азота с мочой сводится к минимуму, поскольку поглощение отфильтрованных кетоновых тел почками становится более полным. Корректировка содержания Na + в моче служит для минимизации потери K + почками и, наряду с изменениями pH мочи, минимизирует вероятность осаждения мочевой кислоты. В ответ на голодание возникает половой диморфизм.
Кетоацидоз является основным признаком распространенных клинических состояний, включая диабетический кетоацидоз, алкогольный кетоацидоз, интоксикацию салицилатами, терапию ингибиторами SGLT2 и диету с достаточным количеством калорий, но с ограничением углеводов. Знание патофизиологии и метаболических последствий кетогенеза имеет решающее значение, учитывая вероятность того, что клиницист может столкнуться с одним из этих состояний.
Введение
Кетоацидоз развивается, когда потребление калорий недостаточно для удовлетворения нормальных метаболических потребностей. Дисбаланс в использовании топлива может привести к кетозу при хронических заболеваниях, при которых анорексия сосуществует с повышенным катаболизмом. Другие причины кетоацидоза включают диабетический кетоацидоз, алкогольный кетоацидоз, интоксикацию салицилатами, терапию ингибиторами SGLT2 и диету с достаточным количеством калорий, но с ограничением углеводов [1-6]. Знание патофизиологии и метаболических последствий кетогенеза имеет решающее значение, учитывая вероятность того, что клиницист может столкнуться с одним из этих состояний. В этом обзоре описаны метаболические изменения, которые происходят, когда полный и в остальном нормальный человек голодает в течение длительного периода времени. Эти изменения можно последовательно разделить на постабсорбционную, глюконеогенную и консервационную белковую фазы.
Постабсорбционная фаза
В первые 24 часа голодания завершение всасывания глюкозы с пищей приводит к падению уровня глюкозы в крови, что сигнализирует об уменьшении циркулирующего инсулина и повышении уровня глюкагона. Глюкагон стимулирует высвобождение глюкозы из запасов гликогена в печени, в то время как снижение уровня инсулина снижает транспорт глюкозы в скелетные мышцы и жировые ткани, обеспечивая достаточное количество глюкозы в крови, доступной для мозга, где она полностью окисляется до углекислого газа и вода [7]. Этот ответ также обеспечивает необходимое топливо для тканей, которые являются исключительно гликолитическими, таких как эритроциты, мозговое вещество почек и костный мозг (рис. 1).
Рисунок 1.
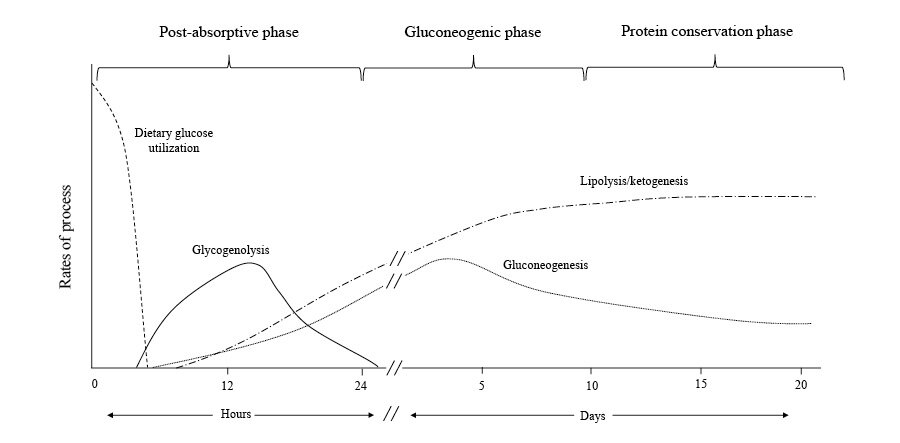
Гликогенолиз в печени происходит из ее типичного запаса в 70 г и обеспечивает около 75% потребности в глюкозе в постабсорбтивной фазе. Глюкозо-6-фосфатаза в печени удаляет фосфатную группу из глюкозо-6-фосфата, образуя свободную глюкозу, которая высвобождается в кровоток для поглощения другими клетками [8]. Гликоген также хранится в скелетных мышцах, но из-за отсутствия глюкозо-6-фосфатазы мышечный гликоген сначала должен метаболизироваться до лактата, который затем высвобождается в кровообращение и повторно синтезируется в глюкозу печенью посредством цикла Кори. Примерно 10–15% оставшейся потребности в глюкозе на этой фазе происходит в результате глюконеогенеза с использованием лактата и пирувата в качестве субстратов. Глицерин, побочный продукт гидролиза триглицеридов, также служит глюконеогенным предшественником [9].
Снижение уровня инсулина активирует липолиз, делая жирные кислоты доступными для использования в качестве альтернативного топлива для скелетных мышц на более поздних стадиях постабсорбционной фазы. При окислении жирных кислот образуется ацетил-КоА, который оказывает ингибирующее действие на пируватдегидрогеназу [10]. Этот эффект гарантирует, что небольшое количество оставшейся глюкозы, поглощенной скелетными мышцами, не подвергается полному окислению в цикле лимонной кислоты, а преимущественно метаболизируется до пирувата и лактата, которые затем превращаются обратно в глюкозу в печени. Мобилизация и окисление жирных кислот в печени обеспечивают энергию для производства глюкозы, поскольку на каждую молекулу глюкозы, синтезируемой в цикле Кори, приходится 4 молекулы АТФ. Раннее использование активности цикла Кори в постабсорбционном состоянии сохраняет белок, избавляя от необходимости в предшественниках аминокислот для глюконеогенеза.
Глюконеогенная фаза
Поскольку запасы гликогена истощаются после 24 часов голодания, пациенты вступают в глюконеогенную фазу, когда значительные количества глюконеогенных предшественников, полученных из аминокислот, добавляются к лактату, пирувату и глицерину, чтобы удовлетворить потребности мозга в глюкозе (рис. 1) . Стойкое снижение уровня инсулина способствует протеолизу в мышцах, обеспечивая необходимый запас субстрата для усиления печеночного глюконеогенеза. Аланин и глутамин являются наиболее распространенными аминокислотами, выделяемыми скелетными мышцами. Несмотря на то, что аланин составляет лишь около 7–10% аминокислотных остатков в скелетных мышцах, на его долю приходится 30–40% аминокислот, высвобождаемых из мышц во время этой фазы [11–13]. Концентрация в плазме аминокислот с разветвленной цепью (лейцина, изолейцина и валина) увеличивается в начале голодания и достигает пика примерно на 5-й день [12]. Эти аминокислоты преимущественно катаболизируются в скелетных мышцах до α-кетокислот путем переаминирования пирувата и служат основным предшественником для образования аланина. Аланин высвобождается скелетными мышцами и попадает в печень; углеродный скелет превращается в глюкозу, а аминогруппа превращается в мочевину и выводится с мочой. Этот аланин-глюкозный цикл переносит аминогруппы аминокислот с разветвленной цепью в печень без повышения уровня аммиака в крови и обеспечивает контрольные точки для ингибирования глюконеогенеза по принципу обратной связи. Например, повышенные концентрации кетокислот оказывают ингибирующее действие на глюконеогенез, уменьшая деградацию аминокислот с разветвленной цепью, тем самым удаляя источник азота для синтеза аланина [14-17]. Кроме того, инсулин оказывает ингибирующее действие на захват аланина печенью, оказывая дополнительный смягчающий эффект на глюконеогенез [18].
Углеродный скелет для синтеза глютамина состоит из таких аминокислот, как глутамат, аспартат, валин и изолейцин. Глутамин служит основным источником энергии для быстрого обновления клеток слизистой оболочки кишечника и клеток иммунной системы. Некоторая часть глутамина, поступающего в кишечник, окисляется лишь частично, чтобы обеспечить дополнительный аланин для печеночного глюконеогенеза [19, 20]. Глутамин также является основным субстратом глюконеогенеза в почках, где производство аммиака в качестве побочного продукта играет важную роль в поддержании кислотно-щелочного баланса [21].
Поток жирных кислот в печень продолжает увеличиваться во время глюконеогенной фазы и направлен в первую очередь на образование кетоновых тел. Увеличение глюкагона приводит к активации 5'-аденозинмонофосфат-активируемого белка, который ингибирует активность ацетил-КоА-карбоксилазы и одновременно активирует малонил-КоА-декарбоксилазу [22, 23] (рис. 2). Снижение уровня малонил-КоА активирует карнитинпальмитоилтрансферазу-I, облегчая проникновение жирных ацильных групп в митохондрии. В норме ацетил-КоА, образующийся в результате β-окисления жирных кислот в печени, подвергается полному окислению в цикле лимонной кислоты с последующим развитием цепи переноса электронов с образованием АТФ. Поскольку печень может производить АТФ только в количестве, равном тому, которое может быть использовано, выработка кетокислот служит путем перетока большого количества вырабатываемого ацетил-КоА [24]. Истощение оксалоацетата из-за усиления глюконеогенеза также способствует кетогенезу, поскольку этот субстрат необходим для входа ацетил-КоА в цикл лимонной кислоты. Кроме того, накопление ацетил-КоА обеспечивает использование пирувата в качестве субстрата для глюконеогенеза, оказывая ингибирующее действие на пируватдегидрогеназу [10].
Рис. 2.
Сохранение белковой фазы
Сохранение белковой фазы характеризуется изменением предпочтений в отношении топлива, что обусловлено необходимостью сохранения глюкозы в крови и защиты белка от постоянной деградации. На этом этапе происходит усиленное производство кетоновых тел, которые используются в качестве топлива для мозга вместо глюкозы [25]. Утилизация глюкозы мозгом падает со 120 г в день в первые 24 часа лишения пищи до примерно 40 г в день после нескольких недель голодания [26, 27]. Распад мышечного белка снижается с 75 до примерно 20 г в день, главным образом, из-за уменьшения высвобождения аланина из мышц [11, 12]. Кетоновые тела непосредственно ингибируют протеолиз мышц и способствуют адаптации к длительному голоданию [14, 28] (рис. 3). Лактат, пируват и глицерин, высвобождаемые во время липолиза, обеспечивают субстрат для оставшегося производства глюкозы печенью. Утилизация глюкозы продолжается в эритроцитах, костном мозге и мозговом веществе почек, которые полагаются исключительно на глюкозу для производства энергии посредством гликолиза. Метаболизм глюкозы в этих исключительно гликолитических тканях ограничивается выработкой пирувата и лактата, которые повторно синтезируются в глюкозу печенью посредством цикла Кори. В ходе этого процесса перерабатывается около 40 г глюкозы в день, и при этом не требуется расщепления белка. Снижение глюконеогенеза в печени сопровождается значительным увеличением глюконеогенеза в почках. Повышенная экстракция глютамина проксимальными канальцами приводит к образованию аммиака, который служит противоанионом для экскреции с мочой солей кетокислоты. [29] (рис. 4). Этот процесс генерирует новый бикарбонат, чтобы компенсировать бикарбонат, расходуемый на буферизацию производства кетокислоты. Когда бикарбонат вводится пациентам, длительно голодающим в количестве, достаточном для коррекции системного ацидоза, экскреция азота с мочой значительно снижается [30, 31]. Этот ответ предполагает, что кислотно-основные факторы не менее важны, если не более важны, чем выработка глюкозы, в стимулировании поглощения глютамина почками.
Рис. 3.
Рис. 4.
Переход от печени к почкам как преобладающему месту глюконеогенеза отражается изменениями продуктов выделения азота с мочой. Вначале наблюдается высокая экскреция мочевины с мочой, которая затем постепенно снижается, при этом преобладающим азотистым продуктом становится аммиак [27]. Хотя эти изменения отражают снижение распада мышечного белка, они также позволяют предотвратить олигиру. Учитывая, что каждая молекула мочевины синтезируется из 2 ионов NH 4 + и 2 HCO 3 - , выведение с мочой NH 4 + в сочетании с β-гидроксибутиратом обеспечивает в 4 раза большее количество осмолей по сравнению с мочевиной [32]. Согласно одному расчету, для обеспечения необходимого количества осмолей (NH 4 -β-гидроксибутират по сравнению с мочевиной) требуется на 75% меньше катаболизма белка для поддержания суточного диуреза 500 мл при концентрации мочи 600 мОсм/кг H 2 O. [32]. В отсутствие жаркой или сухой окружающей среды азотсберегающий эффект длительного голодания может снизить осмотическую экскреторную нагрузку до уровня, требующего выделения только 100–200 мл/день мочи, что позволяет выжить при 200–300 мл/день вырабатываемой воды. путем метаболизма с минимальным дополнительным потреблением воды [33].
В фазе консервации белка производство кетоновых тел печенью в конечном итоге приведет к их равному использованию в мозге, мышцах и почках, за вычетом небольшого количества, выводимого с мочой. В этом устойчивом состоянии голодный кетоз характеризуется концентрацией бикарбоната в плазме примерно 18 мэкв/л, концентрацией β-гидроксибутирата 8–10 ммоль/л и концентрацией глюкозы в плазме от нормальной до низкой. Хотя уровень инсулина в плазме снижается, количество инсулина остается достаточным для предотвращения чрезмерной мобилизации жирных кислот. Кетоновые тела играют важную роль в установлении этого нового равновесия, оказывая прямое стимулирующее действие на высвобождение инсулина в сочетании с прямым ингибирующим действием на липолиз в адипоцитах [16, 28, 34] (рис. 3). Кетокислоты и жирные кислоты постепенно заменяют глюкозу в качестве предпочтительного топлива как для скелетных, так и для сердечных мышц по мере того, как голодание переходит в фазу консервации белка. В конце концов, утилизация свободных жирных кислот становится доминирующей, сохраняя кетокислоты для мозга, поскольку поглощение ацетоацетата мышцами возвращается обратно в кровь в виде β-гидроксибутирата, что означает более сниженный окислительно-восстановительный потенциал в мышечных митохондриях, вторичный по отношению к окислению жирных кислот [33] . Это пониженное состояние также связано со снижением протеолиза мышц, что усиливает азотсберегающий эффект кетокислот в скелетных мышцах [35]. Кэхилл предположил, что предпочтение утилизации кетокислот мозгом напрямую коррелирует с соотношением мозг/тушка у разных видов, поскольку мозг преимущественно использует кетокислоты, а не жирные кислоты, преимущественно используемые тушей [33] . В таблице 1 суммированы другие клинические состояния, характеризующиеся кетоацидозом.
Таблица 1.
Роль почек в голодании
Почки играют решающую роль в устойчивом состоянии, достигаемом на этапе консервации белка. При низких концентрациях в плазме отфильтрованные кетоновые тела полностью реабсорбируются насыщаемыми Na + -связанными монокарбоксилатными переносчиками SMCT1 ( SLC5A8 ) и SMCT2 ( SLC5A12 ) в проксимальных канальцах [36, 37] (рис. 5). Кетонурия развивается по мере повышения уровня в плазме и увеличения фильтруемой нагрузки солей кетокислоты. Потеря связанного с Na + ацетоацетата и β-гидроксибутирата в первые несколько дней голодания приводит к отрицательному балансу Na + и является механизмом, ответственным за быструю первоначальную потерю веса, которая происходит в первые дни полного голодания. [38, 39]. Концентрация Cl- в моче в это время низкая и отражает сокращение объема внеклеточной жидкости. По мере усиления аммиагенеза NH 4 + заменяет Na + в качестве облигатного катиона, сопровождающего экскрецию солей органических кислот. На этом этапе Na + и Cl- в моче слабо отражают повышенную проксимальную реабсорбцию в ответ на сокращение объема (Таблица 2). Снижение дистальной доставки Na + ограничивает потерю K + из организма, даже несмотря на то, что уровни минералокортикоидов в крови повышены. Снижение доставки Na + также снижает дистальную доставку H +.секреция, которая наряду с увеличением NH 4 + в моче делает pH мочи более щелочным, тем самым уменьшая риск осаждения мочевой кислоты [32, 40].
Таблица 2.
Рис. 5.
Концентрация β-гидроксибутирата постепенно увеличивается после длительного голодания. Напротив, пик потерь с мочой приходится на 3–4 день, а затем слегка снижается по мере перехода натощак к фазе консервации белка, что указывает на отсутствие канальцевого максимума для реабсорбции [38, 39, 41]. Точный механизм объяснения этих результатов не определен. Насыщаемый канальцевый секреторный процесс с низкой пропускной способностью, опосредованный переносчиками органических анионов на базолатеральной поверхности канальцев, был предложен в качестве механизма стойкой экскреции β-гидроксибутирата с мочой [42].
Повышенная способность к реабсорбции отфильтрованных кетоновых тел является адаптивной реакцией во время голодания по нескольким причинам. Во-первых, минимизация потерь мочи предотвращает потерю потенциального метаболического топлива, поскольку кетоновые тела обеспечивают значительную часть потребности в калориях во время голодания. При длительном голодании реабсорбция кетоновых тел почками экономит примерно 225 ккал/день, которые в противном случае были бы потеряны с мочой [38]. Во-вторых, реабсорбция кетоновых тел почками оказывает ингибирующее действие на аммиагенез (рис. 5). Инфузия β-гидроксибутирата снижает выработку NH 4+ почками у собак и людей с хроническим метаболическим ацидозом [43-45]. Этот эффект дополняется снижением скорости клубочковой фильтрации и снижением фильтруемой нагрузки Na + [46]. Снижение образования аммиака снижает потребность в поглощении глютамина почками, минимизируя распад белка, потенциально сохраняя до 7 г азота в день [38]. Наконец, реабсорбция и последующее окисление кетоновых тел в регенератах почек потребляют HCO 3 − , тем самым уменьшая степень ацидоза, который в противном случае возникает, если он теряется с мочой в виде соли Na + или K + .
Натрийурез
Потеря веса в первые 1–5 дней голодания колеблется от 1 до 2 кг в сутки и постепенно замедляется в среднем до 0,3 кг в сутки в течение последующих 3 недель. Быстрая первоначальная потеря веса происходит главным образом за счет диуреза соли и воды [47-49]. Повышенная эффективность скелетных мышц замедляет последующую потерю веса за счет снижения калорийности мышечного сокращения [см. 50]. Отрицательный баланс Na + у субъектов, которые голодают в течение нескольких дней, составляет почти 350 ммоль по сравнению с потерей Na + 150 ммоль у субъектов, соблюдающих диету, практически не содержащую Na + [47]. Обязательная потеря Na +из-за повышенного образования и выделения с мочой кетоновых тел, что обусловлено прежде всего натрийуретической реакцией на раннее голодание. Как обсуждалось ранее, величина натрийуреза начинает уменьшаться по мере увеличения аммиагенеза, позволяя NH 4 + заменить Na + в качестве основного катиона мочи. Развитие ацидемии способствует раннему натрийуретическому ответу, поскольку метаболический ацидоз оказывает ингибирующее действие на проксимальную реабсорбцию Na + [51]. Повышение уровня глюкагона и падение уровня инсулина связаны с натрийурезом при раннем голодании. Вливание физиологического уровня глюкагона субъектам, не голодающим, вызывает натрийуретический ответ, аналогичный тому, который наблюдается у голодающих [52]. Снижение уровня инсулина может быть связано с тем, что инсулин обычно стимулирует проксимальную реабсорбцию Na + [53]. Возобновление углеводного питания, даже если диета гипокалорийна, приводит к резкому обращению вспять потери соли и воды и приводит к немедленному увеличению веса [54]. В ряде случаев этот ответ связан с задержкой Na + и развитием клинически определяемого отека. В этом ответе участвуют снижение уровня глюкагона и повышение уровня инсулина. Изокалорическое возобновление питания жиром не дает такого эффекта, тогда как возобновление питания белком вызывает отсроченный и менее выраженный антинатриуретический эффект [54].
Другим потенциальным осложнением возобновления углеводного питания является развитие метаболического алкалоза [55]. Поступление углеводов приводит к прекращению печеночного кетогенеза, в то время как метаболизм периферических кетоновых тел регенерирует HCO 3 - . Хотя этот ответ должен вернуть концентрацию HCO 3 − в плазме почти к норме, у некоторых людей образуется новый бикарбонат, вызывающий развитие легкого метаболического алкалоза. Причиной может быть стойкое увеличение проксимального аммиагенеза с продолжающейся повышенной экскрецией кислоты почками вследствие гипертрофии канальцев. Метаболизм сохраненных анионов кетокислот возвращает концентрацию HCO 3 − выше нормальной, поскольку почки NH 4 +выведение превышает скорость выведения кетокислоты. Задержка Na + после поступления углеводов наряду с повышенным уровнем инсулина увеличивает реабсорбционную способность HCO3- проксимальных канальцев и обеспечивает механизм поддержания алкалоза [53]. Метаболический алкалоз при возобновлении питания обычно протекает в легкой форме и спонтанно проходит через несколько дней после возобновления питания по мере снижения аммиагенеза.
Гомеостаз калия во время голодания
У субъектов, голодающих в течение 1 недели, дефицит К + составляет примерно 300 ммоль [56, 57]. Этот ранний калийуретический эффект обусловлен сочетанием повышенных уровней циркулирующего альдостерона из-за сокращения объема внеклеточной жидкости с увеличением дистальной доставки Na + вторично по отношению к нереабсорбируемому анионному эффекту экскреции солей кетокислоты. После нескольких дней голодания степень экскреции K + почками снижается до низкой скорости, составляя в среднем 19 мэкв/сут за 1 месяц голодания. Это снижение обусловлено снижением доставки Na + в дистальный отдел нефрона, поскольку NH4 + все чаще заменяет Na +.в качестве облигатного катиона для экскреции солей кетокислот. Кроме того, измененная функция канальцев в дистальных отделах нефрона, вторичная по отношению к эффектам ангиотензина II, участвует в минимизации секреции K + [58] (Рис. 6). Эти эффекты позволяют повышенным уровням альдостерона участвовать в максимальном сохранении Na + и Cl − без усугубления дефицита общего содержания K + в организме. Концентрация K + в плазме обычно снижается, но редко падает ниже 3,0 мэкв/л при длительном голодании.
Рис. 6.
Мочевая кислота
В норме в день выделяется 400–450 мг мочевой кислоты [47]. рН мочи снижается на ранних стадиях голодания, поскольку развивающийся ацидоз стимулирует секрецию Н + в дистальных отделах нефрона. Этот эффект создает фактор риска преципитации мочевой кислоты, поскольку растворимость мочевой кислоты в кислой моче плохая, учитывая pKa примерно 5,7. Во время голодания этот риск смягчается несколькими факторами. Во-первых, первоначальный диурез голодания увеличивает объем мочи и помогает поддерживать концентрацию мочевой кислоты в концентрации, меньшей, чем произведение ее растворимости. Во-вторых, по мере увеличения циркулирующих уровней β-гидроксибутирата экскреция мочевой кислоты в мочу снижается, поскольку оба вещества конкурируют за одни и те же места транспорта в почках [59]. В-третьих, последующее снижение экскреции Na + с мочой, сопровождающееся прогрессивным увеличением экскреции NH4 + с мочой, служит для смягчения падения pH мочи [40].
Половой диморфизм при голодном кетозе
Половые различия в метаболизме субстратов могут объяснить более быстрое развитие кетоза натощак у женщин [60, 61]. Хотя мужчины и женщины не различаются по проценту энергии, получаемой из углеводов или жиров в состоянии покоя, женщины используют больший процент жирных кислот в качестве основного энергетического субстрата после упражнений, голодания и других метаболических стрессоров по сравнению с мужчинами [62]. Эстрогены могут объяснить эту разницу, поскольку есть данные, позволяющие предположить, что эстрогены способствуют липолизу, усиливают активность вегетативных нервов и регулируют ключевые ферменты в липолитических путях [63]. Кроме того, у женщин липолиз опосредуется β-адренергическими рецепторами, тогда как у мужчин происходит стимуляция не только β-адренорецепторов, но и α-адренорецепторов, что ослабляет липолиз [64]. Важно отметить, что женщины имеют повышенную восприимчивость к кетоацидозу из-за усиленного липолиза, что подвергает их большему риску развития эугликемического кетоацидоза после терапии SGLT2i [2].
Уровни глюкагона выше у женщин, не страдающих ожирением, чем у мужчин, что способствует большей степени кетоза натощак [65]. Усиление липолиза приведет к высвобождению большего количества глицерина, заменяющего аланин в качестве предпочтительного глюконеогенного субстрата. Уровни циркулирующих аминокислот, включая аланин, снижаются у голодающих женщин по сравнению с мужчинами. Изменения в предпочтении субстрата могут представлять собой эволюционную адаптацию, призванную облегчить передачу аминокислот от матери к развивающемуся плоду, а не использовать их матерью для глюконеогенеза.
Половые различия в кетогенной реакции на голодание исчезают с увеличением массы тела [66]. Эта утрата половых различий может быть связана как с более высоким базальным уровнем инсулина, так и с уровнем инсулина натощак, типичным для ожирения, поскольку максимальная мобилизация жирных кислот происходит при снижении уровня инсулина.
Заключение
Организм адаптируется к длительному голоданию, сохраняя азот, поскольку мозг все чаще использует кетокислоты, избавляясь от потребности в глюкозе. Изменения в использовании топлива уменьшают потребность в мобилизации аминокислот из мышц, тем самым обеспечивая возможность сохранения белка. Системная адаптация к голоданию отражается изменениями в биохимическом профиле мочи. Замечательная способность организма адаптироваться к длительному голоданию имела решающее значение для выживания первобытного человека. Очень важно отметить, что большая часть оригинальной работы по кетозу, упомянутой здесь и выполненной Кэхиллом и его коллегами, проводилась на людях, страдающих ожирением. Время наступления фаз и стадий кетотического процесса может различаться у худощавых людей и/или у людей с отсутствием ожирения. Понимание этих процессов может дать клиницисту лучшее понимание многих клинических состояний, характеризующихся кетоацидозом.
Рекомендованная литература
1. Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med. 2015;373(6):548–59.
2. Palmer BF, Clegg DJ. Euglycemic ketoacidosis as a complication of SGLT2 inhibitor therapy. Clin J Am Soc Nephrol. 2021 Feb 9.
3. Palmer BF, Clegg DJ. Electrolyte disturbances in patients with chronic alcohol-use disorder. N Engl J Med. 2017;377(14):1368–77.
4. Metzger BE, Ravnikar V, Vileisis RA, Freinkel N. “Accelerated starvation” and the skipped breakfast in late normal pregnancy. Lancet. 1982;1:588–92.
5. Palmer BF, Clegg DJ. Salicylate toxicity. N Engl J Med. 2020;382(26):2544–55.
6. Shah P, Isley WL. Ketoacidosis during a low-carbohydrate diet. N Engl J Med. 2006;354(1):97–8.
7. Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab. 2011;13(Suppl 1):118–25.
8. Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J. 2002;362(Pt 3):513–32.
9. Wang Y, Kwon H, Su X, Wondisford FE. Glycerol not lactate is the major net carbon source for gluconeogenesis in mice during both short and prolonged fasting. Mol Metab. 2020;31:36–44.
10. BehalBuxton RD, Robertson J, Olson M. Regulation of the pyruvate dehydrogenase multienzyme complex. Annu Rev Nutr. 1993;13:497–520.
11. Felig P. Amino acid metabolism in man. Annu Rev Biochem. 1975;44:933–55.
12. Felig P, Owen OE, Wahren J, Cahill GF Jr. Amino acid metabolism during prolonged starvation. J Clin Invest. 1969;48(3):584–94.
13. Felig P, Wahren J, Sherwin R, Palaiologos G. Amino acid and protein metabolism in diabetes mellitus. Arch Intern Med. 1977;137(4):507–13.
14. Thompson JR, Wu G. The effect of ketone bodies on nitrogen metabolism in skeletal muscle. Comp Biochem Physiol B. 1991;100(2):209–16.
15. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60(1):143–87.
16. Henry RR, Brechtel G, Lim KH. Effects of ketone bodies on carbohydrate metabolism in non-insulin-dependent (type II) diabetes mellitus. Metab Clin Exp. 1990;39(8):853–8.
17. Hanson PJ, Parsons DS. Factors affecting the utilization of ketone bodies and other substrates by rat jejunum: effects of fasting and of diabetes. J Physiol. 1978;278:55–67.
18. Qian K, Zhong S, Xie K, Yu D, Yang R, Gong DW. Hepatic ALT isoenzymes are elevated in gluconeogenic conditions including diabetes and suppressed by insulin at the protein level. Diabetes Metab Res Rev. 2015;31(6):562–71.
19. Marliss EB, Aoki TT, Pozefsky T, Most AS, Cahill GF. Muscle and splanchnic glutmine and glutamate metabolism in postabsorptive andstarved man. J Clin Invest. 1971;50(4):814–7.
20. Watford M. Glutamine metabolism in rat small intestine: synthesis of three-carbon products in isolated enterocytes. Biochim Biophys Acta. 1994;1200(1):73–8.
21. Ross BD, Espinal J, Silva P. Glucose metabolism in renal tubular function. Kidney Int. 1986;29(1):54–67.
22. Angin Y, Beauloye C, Horman S, Bertrand L. Regulation of carbohydrate metabolism, lipid metabolism, and protein metabolism by AMPK. Exp Suppl. 2016;107:23–43.
23. Saha AK, Ruderman NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem. 2003;253(1–2):65–70.
24. Jungas RL, Halperin ML, Brosnan JT. Quantitative analysis of amino acid oxidation and related gluconeogenesis in humans. Physiol Rev. 1992;72(2):419–48.
25. Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF. Brain metabolism during fasting. J Clin Invest. 1967;46(10):1589–95.
26. Cahill GF Jr, Owen OE. Starvation and survival. Trans Am Clin Climatol Assoc. 1968;79:13–20.
27. Cahill G. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22.
28. Madison LL, Mebane D, Unger RH, Lochner A. The hypoglycemic action of ketones. II. Evidence for a stimulatory feedback of ketones on the pancreatic beta cells. J Clin Invest. 1964;43(3):408–15.
29. Halperin ML, Jungas RL. Metabolic production and renal disposal of hydrogen ions. Kidney Int. 1983;24(6):709–13.
30. Féry F, Balasse EO. Differential effects of sodium acetoacetate and acetoacetic acid infusions on alanine and glutamine metabolism in man. J Clin Invest. 1980;66(2):323–31.
31. Hannaford MC, Leiter LA, Josse RG, Goldstein MB, Marliss EB, Halperin ML. Protein wasting due to acidosis of prolonged fasting. Am J Physiol. 1982;243(3):E251–6.
32. Kamel SK, Lin SH, Cheema-Dhadli S, Marliss EB, Halperin ML. Prolonged total fasting: a feast for the integrative physiologist. Kidney Int. 1998;53(3):531–9.
33. Cahill GF. President’s address. Starvation. Trans Am Clin Climatol Assoc. 1983;94:1–21.
34. Björntorp P, Scherstén T. Effect of beta-hydroxybutyrate on lipid mobilization. Am J Physiol. 1967;212(3):683–7.
35. Aoki TT, Finley RJ, Cahill GF. The redox state and regulation of amino acid metabolism in man. Biochem Soc Symp. 1978;43:17–29.
36. Gopal E, Fei YJ, Sugawara M, Miyauchi S, Zhuang L, Martin P, Expression of slc5a8 in kidney and its role in Na(+)-coupled transport of lactate. J Biol Chem. 2004;279(43):44522–32.
37. Gopal E, Umapathy NS, Martin PM, Ananth S, Gnana-Prakasam JP, Becker H, Cloning and functional characterization of human SMCT2 (SLC5A12) and expression pattern of the transporter in kidney. Biochim Biophys Acta. 2007;1768(11):2690–7.
38. Sapir DG, Owen OE. Renal conservation of ketone bodies during starvation. Metab Clin Exp. 1975;24:23–33.
39. Owen OE, Caprio S, Reichard GA Jr, Mozzoli MA, Boden G, Owen RS. Ketosis of starvation: a revisit and new perspectives. Clin Endocrinol Metab. 1983 Jul;12(2):359–79.
40. Batlle DC, von Riotte A, Schlueter W. Urinary sodium in the evaluation of hyperchloremic metabolic acidosis. N Engl J Med. 1987;316(3):140–4.
41. Barac-Nieto M. Renal reabsorption and utilization of hydroxybutyrate and acetoacetate in starved rats. Am J Physiol. 1986;251(2 Pt 2):F257–65.
42. Wang K, Kestenbaum B. Proximal tubular secretory clearance: a neglected partner of kidney function. Clin J Am Soc Nephrol. 2018;13(8):1291–6.
43. Desir G, Bratusch-Marrain P, DeFronzo RA. Effect of hyperketonemia on renal ammonia excretion in man. Metab Clin Exp. 1986;35(8):736–43.
44. Sherwin RS, Hendler RG, Felig P. Effect of ketone infusions on amino acid and nitrogen metabolism in man. J Clin Invest. 1975;55(6):1382–90.
45. Lemieux G, Pichette C, Vinay P, Gougoux A. Cellular mechanisms of the antiammoniagenic effect of ketone bodies in the dog. Am J Physiol. 1980;239(5):F420–6.
46. Halperin ML, Cheema-Dhadli S. Renal and hepatic aspects of ketoacidosis: a quantitative analysis based on energy turnover. Diabetes Metab Rev. 1989;5(4):321–36.
47. Kerndt P, Naughton J, Driscoll C, Loxterkamp D. Fasting: the history, pathophysiology and complications. West J Med. 1982;137(5):379–99.
48. North KA, Lascelles D, Coates P. The mechanisms by which sodium excretion is increased during a fast but reduced on subsequent carbohydrate feeding. Clin Sci Mol Med. 1974;46(4):423–32.
49. Sigler MH. The mechanism of the natriuresis of fasting. J Clin Invest. 1975;55(2):377–87.
50. Palmer BF, Clegg DJ. Non-shivering thermogenesis as a mechanism to facilitate sustainable weight loss. Obes Rev. 2017;18(8):819–31.
51. Aronson PS, Giebisch G. Effects of pH on potassium: new explanations for old observations. J Am Soc Nephrol. 2011;22(11):1981–9.
52. Saudek CD, Boulter PR, Arky RA. The natriuretic effect of glucagon and its role in starvation. J Clin Endocrinol Metab. 1973;36(4):761–5.
53. Ruiz OS, Qiu YY, Cardoso LR, Arruda JA. Regulation of the renal Na-HCO3 cotransporter: IX. Modulation by insulin, epidermal growth factor and carbachol. Regul Pept. 1998;77(1–3):155–61.
54. Veverbrants E, Arky RA. Effects of fasting and refeeding. I. Studies on sodium, potassium and water excretion on a constant electrolyte and fluid intake. J Clin Endocrinol Metab. 1969;29(1):55–62.
55. Stinebaugh BJ, Schloeder FX. Glucose-induced alkalosis in fasting subjects. Relationship to renal bicarbonate reabsorption during fasting and refeeding. J Clin Invest. 1972;51(6):1326–36.
56. Drenick EJ, Blahd WH, Singer FR, Lederer M. Body potassium content in obese subjects and potassium depletion during prolonged fasting. Am J Clin Nutr. 1966;18(4):278–85.
57. Lin SH, Cheema-Dhadli S, Gowrishankar M, Marliss EB, Kamel KS, Halperin ML. Control of excretion of potassium: lessons from studies during prolonged total fasting in human subjects. Am J Physiol. 1997;273(5):F796–800.
58. Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis: core curriculum 2019. Am J Kidney Dis. 2019;74(5):682–95.
59. Goldfinger S, Klinenberg E, Seegmiller JE. Renal retention of uric acid induced by infusion of beta-hydroxybutyrate and acetoacetate. N Engl J Med. 1965;272:351–5.
60. Marinou K, Adiels M, Hodson L, Frayn KN, Karpe F, Fielding BA. Young women partition fatty acids towards ketone body production rather than VLDL-TAG synthesis, compared with young men. Br J Nutr. 2011;105(6):857–65.
61. Bloom W. Fasting ketosis in obese men and women. Transl Res. 1962;59(4):605–12.
62. Hedrington MS, Davis SN. Sexual dimorphism in glucose and lipid metabolism during fasting, hypoglycemia, and exercise. Front Endocrinol. 2015;6:61.
63. Davis SN, Galassetti P, Wasserman DH, Tate D. Effects of gender on neuroendocrine and metabolic counterregulatory responses to exercise in normal man. J Clin Endocrinol Metab. 2000;85(1):224–30.
64. Palmer BF, Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. 2015;402:113–9.
65. Merimee TJ, Misbin RI, Pulkkinen AJ. Sex variations in free fatty acids and ketones during fasting: evidence for a role of glucagon. J Clin Endocrinol Metab. 1978;46(3):414–9.
66. Kekwick A, Pawan GL, Chalmers TM. Resistance to ketosis in obese subjects. Lancet. 1959;2(7113):1157–9.