Недавние исследования показали, что развитие НАЖБП связано с накоплением липидов, окислительным стрессом, стрессом эндоплазматического ретикулума и липотоксичностью.
Ранние исследования предполагают, что ИР и стеатоз печени из-за избытка жирных кислот являются «первым ударом», тогда как гепатоциты в конечном итоге подвергаются повреждению, воспалению, фиброзу и другим патологическим изменениям из-за окислительного стресса и перекисного окисления липидов, образуя «второй удар» [13].
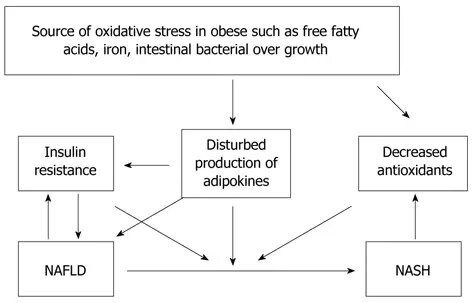
Рисунок 1 Окислительный стресс является одним из наиболее популярных предложенных механизмов гепатоклелярного повреждения и возможный источник окисляющего стресса следующим образом: увеличение свободного содержания жирных кислот, железа, кишечника бактериального роста. NASH: Неалкогольная стеатогепатит; NAFLD: Неалкогольная жирная болезнь печени.
Сегодня широко признано, что теория «множественного удара» основана на теории «второго удара», которая включает различные факторы, такие как окислительный стресс, стресс эндоплазматического ретикулума (ЭР) и липотоксичность [14].
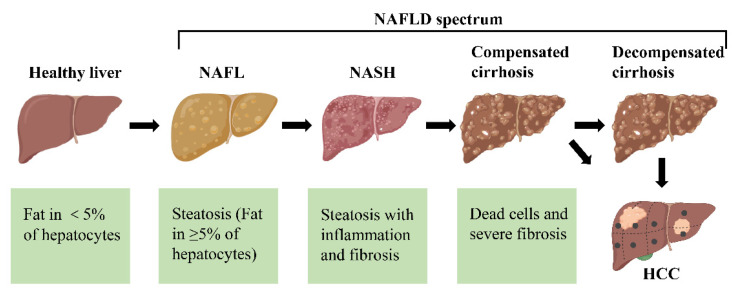
Термин НАЖБП охватывает широкий спектр состояний, от простого накопления жира («жировая печень» или стеатоз) до стеатогепатита (НАЖБП), фиброза и цирроза с его клиническими последствиями.
Несмотря на высокую распространенность НАЖБП в общей популяции, у подавляющего большинства пациентов наблюдается простой стеатоз, который не связан с ухудшением выживаемости.
Только у 5–10% пациентов с диагнозом НАЖБП разовьется НАЖБП, и у 30% из них разовьется цирроз.
Рис. 2– Гипотеза множественного удара для развития НАЖБП. Сокращения: СЖК, свободные жирные кислоты; DNL, липогенез de novo;ЛПОНП, липопротеины очень низкой плотности; CH, холестерин; TNF-α, фактор некроза опухоли альфа; IL-6, интерлейкин 6; TG, триглицериды;ROS, активные формы кислорода; ER, эндоплазматический ретикулум; UPR, развернутый белковый ответ; LPS, липополисахарид; НАЖБП, неалкогольная жировая болезнь печени; NASH, неалкогольный стеатогепатит.
Диетические и экологические факторы, вместе с ожирением, приводят к повышению уровня жирных кислот (СЖК) и холестерина (Х), развитию резистентности к инсулину, пролиферации и дисфункции адипоцитов и изменениям в микробиоме кишечника.
Инсулинорезистентность действует на жировую ткань, ухудшая дисфункцию адипоцитов, вызывает липолиз и высвобождение адипокинов и провоспалительных цитокинов, таких как ФНО-α и ИЛ-6, которые также способствуют поддержанию состояния инсулинорезистентности.
В печени инсулинорезистентность усиливает DNL.
Увеличенный поток свободных жирных кислот в печени, который возникает из-за вышеуказанных процессов и из-за измененной активности микробиома кишечника, приводит к двум различным ситуациям: синтез и накопление триглицеридов (ТГ) и «токсичных» уровней жирных кислот, свободного холестерина и других липидных метаболитов, которые вызывают дисфункцию митохондрий с окислительным стрессом и выработкой ROS и стресса эндоплазматического ретикулума (ЭР) с активацией UPR, все это приводит к воспалению печени.
Также проницаемость тонкой кишки может быть усилена с последующим повышением циркулирующих уровней молекул, которые способствуют активации инфламмасомы и стресса ER, таких как ЛПС, и высвобождению провоспалительных цитокинов.
Генетические факторы или эпигенетические модификации влияют на содержание жира в гепатоцитах, ферментативные процессы и воспалительную среду печени, тем самым влияя на риск прогрессирования воспаления и фиброза (НАСГ) или сохранения стабильной стадии заболевания (НАЖБП).
В настоящее время нет клинически одобренных препаратов для лечения НАЖБП, и лечение в основном заключается в диете и физических упражнениях для изменения образа жизни [15].
Гепатостеатоз при НАЖБП вызван избыточным синтезом триглицеридов (ТГ) в гепатоцитах, при этом 60% субстрата для этого синтеза поступает из белой жировой ткани (БЖТ), 26% — из липогенеза de novo (DNL) и 15% — из потребления диеты с высоким содержанием жиров и/или сахара [19,20,21].
Интересно, что у мышей, получавших глюкозу, уровни портального эндотоксина не отличались от контрольных, что еще раз подтверждает гипотезу о том, что источник калорий, а а не количество, может иметь решающее значение для развития НАЖБП.
Аномальное накопление липидов при НАЖБП.
Увеличение накопления липидов в печени происходит из-за поглощения большого количества свободных жирных кислот (СЖК), синтезированных печенью триглицеридов из белой жировой ткани (БЖТ), продуктов с высоким содержанием жиров и сахара, а также липогенеза de novo (DNL).
Инсулинорезистентность играет жизненно важную роль в этом процессе. Инсулинорезистентность способствует усвоению глюкозы и усиливает липолиз БЖТ. Это приводит к активации пути DNL. Сокращения: ChREBP, белок, связывающий элемент углеводного ответа; SREBP-1c, белок, связывающий элемент стерола 1c; ACC, ацетил-КоА-карбоксилаза; FAS, синтаза жирных кислот.
Инсулин обладает антилиполитическим эффектом, опосредует хранение ТГ в жировой ткани и способствует этерификации и хранению жирных кислот [22]. Таким образом, инсулинорезистентность (ИР) становится ключевым терапевтическим фактором при НАЖБП.
Жирная кислота в основном хранится в липидных каплях WAT в виде ТГ [23]. Липидные капли в клетках долгое время использовались как относительно ленивый липидный резервуар [24].
Они действуют как батарея, чтобы хранить избыточную энергию и высвобождать ее при необходимости. В состоянии ИР антилиполитический эффект инсулина уменьшается, и WAT расщепляется, что приводит к большому высвобождению свободных жирных кислот (СЖК) [25]. Затем избыток СЖК хранится в печени в виде ТГ, образуя липидные эктопические отложения и вызывая НАЖБП [26].
DNL является ключевым путем, который способствует накоплению липидов и тесно связан с ИР [27]. DNL модулируется белком 1c, связывающим регуляторный элемент стерола (SREBP-1c), и белком, связывающим элемент углеводного ответа (ChREBP) [28,29]. IR активирует SREBP-1c для стимулирования DNL в гепатоцитах [30,31].
Повышенная концентрация глюкозы активирует ChREBP для регулирования экспрессии ацетил-КоА-карбоксилазы (ACC) и синтазы жирных кислот (FAS), тем самым стимулируя DNL в гепатоцитах [32,33,34].
С эпидемией ожирения мы обнаружили, что диетические факторы имеют решающее значение для развития НАЖБП [35,36,37].
Исследование показало, что диета с высоким содержанием жиров (HFD) сама по себе приводит к ожирению, IR и некоторой степени жировой печени с небольшим воспалением и фиброзом, тогда как диета с добавлением фруктозы увеличивает экспрессию генов фиброза печени, воспаления, стресса ER и апоптоза адипоцитов. [38].
Кроме того, модели животных и исследования на людях показали, что фруктоза имеет селективный метаболизм в печени и запускает реакции печени на стресс, включая активацию c-Jun N-терминальной киназы (JNK) и IR, что способствует накоплению жира в печени, что приводит к усилению липогенеза и нарушению окисления жирных кислот (FAO), вызывая воспаление печени и фиброз печени [39,40,41,42].
Это говорит о том, что фруктоза в составе рациона является важным фактором риска развития НАЖБП в НАСГ.
Окислительный стресс
Обычно DNL преобразует избыток углеводов в жирные кислоты. Поэтому эти жирные кислоты этерифицируются с образованием триглицеридов (ТГ), которые хранятся в гепатоцитах. В периоды дефицита энергии ТГ обеспечивает организм энергией посредством β-окисления [43]. Увеличение свободных жирных кислот в печени из-за различных причин приводит к повреждению β-окисления и дисфункции митохондрий, что приводит к воспалению, которое приводит к окислительному стрессу (рисунок 3) [44].
Активные формы кислорода (ROS) являются важными медиаторами воспалительной реакции [45,46].
Пероксисома является первым ферментом системы β-окисления жирных кислот. Рецептор, активируемый пролиферацией пероксисом α (PPARα), регулирует активность трех взаимосвязанных систем окисления жирных кислот в печени, а именно митохондриального и пероксисомального β-окисления и микросомального ω-окисления [47].
Устойчивая активация PPARα может облегчить НАЖБП за счет повышения уровня FAO и снижения уровня ROS [48,49,50]. Однако многие исследования показали, что чрезмерная активация PPARα повышает уровень FAO в печени, а также приводит к чрезмерному сжиганию энергии в печени, непропорционально увеличивая H2O2 и вызывая воспалительную реакцию [51,52,53]. У пациентов с НАЖБП наблюдаются ультраструктурные повреждения митохондрий, снижение активности комплекса дыхательной цепи и нарушение синтеза АТФ [54]. Митохондрии играют очень важную роль в обеспечении жирных кислот и энергии, но в этом процессе также вырабатывается большое количество АФК, что является одним из основных источников АФК в клетках [55]. ACC катализирует DNL и регулирует митохондриальную FAO [56]. DNL усиливает гликолитическую активность, что приводит к повышению уровня пирувата и ацетил-КоА. СЖК пересекают внутреннюю митохондриальную мембрану через карнитинпальмитоилтрансферазу 1 (CPT1) [57]. Нарушение митохондриального β-окисления происходит, когда снижается транспорт жирных кислот в митохондрии [58,59]. В митохондриях ацил-КоА преобразуется в ацетил-КоА путем β-окисления, а затем входит в цикл трикарбоновых кислот (ЦТК) для обеспечения энергии. Более конкретно, митохондриальная дисфункция вызвана повреждением цепи переноса электронов (ETC). Компоненты митохондриальной дыхательной цепи чрезмерно восстанавливаются электронами, которые затем ненормально реагируют с кислородом, что приводит к увеличению ROS [60]. Кроме того, ROS окисляют жировые отложения, высвобождая липидные перекиси, которые повреждают гепатоциты. В гепатоцитах ROS и липидные перекиси еще больше повреждают дыхательную цепь, напрямую или косвенно вызывая окислительное повреждение митохондриального генома, что также приводит к образованию большего количества ROS, тем самым создавая порочный круг [61,62].
Стресс эндоплазматического ретикулума (ER)
ER-стресс — это защитная реакция, которая восстанавливает гомеостаз белков путем активации реакции развернутого белка (UPR) [63].
Однако, когда активация UPR не способствует выживанию клеток, клетки активируются проапоптотическим путем стресса ER, что в конечном итоге приводит к гибели клетки (рисунок 4) [64].
Мембрана ER состоит из небольшого количества холестерина и сложных сфинголипидов [65].
Эта свободная упаковка липидов мембраны ER облегчает синтез новых липидов и транспорт белков. Липогенез является основным метаболическим путем, на который влияет стресс ER [66].
Последние данные свидетельствуют о том, что ER присутствует как при развитии стеатоза печени, так и при прогрессировании НАЖБП [67]. Сообщалось, что нарушенный гомеостаз ER обнаружен в печени пациентов с НАЖБП [68]. Этот результат свидетельствует о том, что стресс ER тесно связан с НАЖБП.
Липидный состав различных мембран различается по всей клетке. Данные о липидном составе выражены в процентах от общего количества фосфолипидов (PL) у млекопитающих (синий) и дрожжей (светло-голубой). В качестве меры содержания стеролов также включено молярное отношение холестерина (CHOL; у млекопитающих) и эргостерола (ERG; у дрожжей) к фосфолипидам. Основная панель показывает место синтеза основных фосфолипидов (синий) и липидов, которые участвуют в сигнальных путях и путях распознавания органелл (красный). Следует понимать, что уровни сигнальных и распознающих липидов значительно ниже 1% от общего количества фосфолипидов, за исключением церамида (Cer). Основные глицерофосфолипиды, собранные в эндоплазматическом ретикулуме (ЭР), — это фосфатидилхолин (PtdCho; PC), фосфатидилэтаноламин (PtdEtn; PE), фосфатидилинозитол (PtdIns; PI), фосфатидилсерин (PtdSer; PS) и фосфатидная кислота (PA). Кроме того, ЭР синтезирует Cer, галактозилцерамид (GalCer), холестерин и эргостерол. И ЭР, и липидные капли участвуют в синтезе стерилового эфира и триацилглицерола (TG). Просвет Гольджи является местом синтеза сфингомиелина (SM), сложных гликосфинголипидов (GSL) и синтеза дрожжевого инозитолсфинголипида (ISL). PtdCho также синтезируется в аппарате Гольджи и может быть связан с секрецией белка на уровне его предшественника диацилглицерина (ДАГ). Примерно 45% фосфолипидов в митохондриях (в основном PtdEtn, PA и кардиолипин (CL)) автономно синтезируются органеллой. BMP (бис(моноацилглицеро)фосфат) является основным фосфолипидом во внутренних мембранах поздних эндосом26. PI(3,5)P2, фосфатидилинозитол-(3,5)-бисфосфат; PI(4,5)P2, фосфатидилинозитол-(4,5)-бисфосфат; PI(3,4,5)P3, фосфатидилинозитол-(3,4,5)-трифосфат; PI4P, фосфатидилинозитол-4-фосфат; R, остальные липиды; S1P, сфингозин-1-фосфат.
.
По мере перемещения липидов из эндоплазматического ретикулума (ЭР) в аппарат Гольджи, плазматическую мембрану и в эндосомы внутренние липидные транспортеры определяют распределение фосфолипидов через бислой. a В ЭР было продемонстрировано неспецифическое трансбилисное равновесие фосфолипидов, и мембрана демонстрирует почти симметричное распределение липидов между листками бислоя. b В аппарате Гольджи АТФазы P4 перемещают фосфатидилсерин (PtdSer; PS) и фосфатидилэтаноламин (PtdEtn; PE) на цитозольную поверхность. Сфингомиелин (СМ) вырабатывается синтазой СМ из церамида (Cer) на люменальной стороне. Ни фосфатидилхолин (PtdCho; PC), ни молекулы СМ, которые находятся в люменальной поверхности, не транспортируются на цитозольную поверхность. Таким образом, асимметрия в аппарате Гольджи создается специфическим транспортом PtdSer и PtdEtn и отсутствием транспорта SM и PtdCho. При синтезе SM PtdCho преобразуется в диацилглицерол (DAG), который свободно уравновешивается через бислои. DAG может служить субстратом для изофермента холинфосфотрансферазы, продуктом которого является SM. c На плазматической мембране P4 АТФазы транспортируют PtdSer и PtdEtn к цитозольной поверхности, при небольшом или отсутствующем транспорте PtdCho или SM к цитозольной поверхности в базальных условиях. Это гомеостатическое распределение может быть нарушено активацией скрамблазы и/или ингибированием P4 АТФаз. d Внутри эндосом флуоресцентные PtdCho и SM и гликосфинголипиды (GSL) ограничены люменальным листком из-за отсутствия специфических транспортных механизмов. P4 АТФазы рециркулируются через эндосомы123.