Заболевание, связанное с избыточным накоплением меди в тканях, особенно в печени, в связи с наследственным аутосомно-рецессивным дефектом белка, транспортирующего медь, локализованного в мембране гепатоцитов. Данный дефект вызван мутацией гена ATP7B, который расположен на 13 хромосоме (выявлено >500 мутаций, большинство больных являются комплексными гетерозиготами, т. е. у них присутствуют 2 разные мутации). Результатом этого является нарушение выведения меди с желчью и ее накопление в печени, головном мозге, почках и роговице, что приводит к повреждению этих органов.
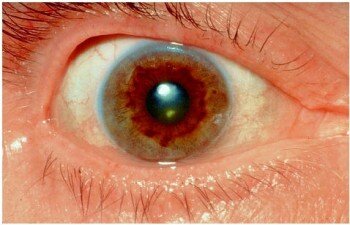
Первые симптомы обычно появляются в возрасте 5–40 лет, редко (≈3 %) в более позднем возрасте. Клиническая картина очень разнообразна и может включать в себя различные системы и органы. При отсутствии лечения болезнь прогрессирует; может возникнуть острая печеночная недостаточность, которая характеризуется высокой смертностью без срочной трансплантации печени; ранняя диагностика и лечение облегчает симптомы (напр. вызывает регресс глазных изменений) и предотвращает осложнения.