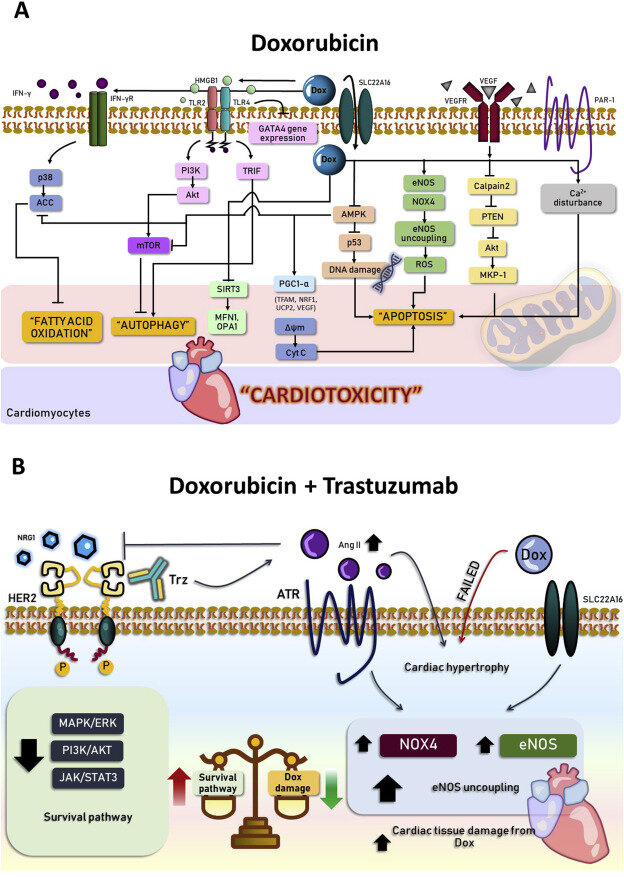
2. Влияние доксорубицина на кардиотоксичность: отчеты об исследованиях in vitro
Хотя основной механизм, участвующий в вызванной Доксом сердечной дисфункции, не ясен, накопленные данные исследований in vitro указывают на то, что вызванная Доксом кардиотоксичность является результатом повышенного окислительного стресса, апоптоза и митохондриальной дисфункции ( Goyal et al., 2016; Liu et al., 2018; Ni et al., 2019). Это нарушение обусловлено ингибирование 5' АМФ-активируемой протеинкиназы (АМПК) и подавление пролифераторами пероксисом рецептор, активируемый гамма коактиватор 1-Альфа (PGC1-α) в результате чего вниз по течению сигнализации (ядерного респираторного фактора 1, NRF1; митохондриальный транскрипционный фактор а, TFAM; и митохондриальных разобщающих белков, UCP2), а также снижение клеточной жизнеспособности кардиомиоцитов (Лю и соавт., 2018). В сердце митохондрии играют важную роль в образовании аденозинтрифосфата (АТФ), и АМПК/ПГК1-α играет центральную роль в митохондриальном энергетическом гомеостазе как при физиологических, так и при патологических состояниях ( Herzig and Shaw, 2018 ). Ингибирование AMPK/PGC1-α Доксом было предложено в качестве одной из основных причин сердечной дисфункции ( Liu et al., 2018). Кроме того, регуляция энергетического метаболизма сердца связана с интерфероном гамма (IFNy) ( Ferreira et al., 2014; Ni et al., 2019). В недавнем исследовании сообщалось, что лечение Доксом стимулировало высвобождение IFNy в кардиомиоцитах ( Levick and Goldspink, 2014 ). В первичных кардиомиоцитах, обработанных Доксом, IFNy подавлял ось AMPK / Acety-CoA карбоксилазы ( ACC) через P38-зависимую ветвь, вызывая нарушенное перепрограммирование метаболизма жирных кислот и нарушенную запасную дыхательную способность митохондрий (Ni et al., 2019).
Кроме того, активация протеазы-активированного рецептора 1 (PAR-1) была предложена в качестве другого механизма, ответственного за вызванную Доксом митохондриальную дисфункцию ( Antoniak et al., 2018). PAR-1-это рецептор, связанный с G-белком, который участвует в повреждении сердца и неблагоприятном ремоделировании сердца ( Pawlinski et al., 2007). Antoniak et al. нашел, что по номиналу-1 -/- кардиомиоциты демонстрировали более низкие уровни оксидативного стресса, апоптоза и митохондриальной дисфункции, чем PAR-1 +/+ после введения Докса ( Antoniak et al., 2018). Кроме того, индуцированный Доксом апоптоз был связан с нарушениями Ca 2+ (Wallace, 2007). Ингибирование Ca 2 + зависимой сигнализации устраняло зависимое от PAR-1 снижение изменений мембранного потенциала митохондрий (Δψm) ( Antoniak et al., 2018). Митохондриальная дезацетилаза sirtuin 3 ( SIRT3) играет важную роль во многих биологических процессах, включая клеточный метаболизм, апоптоз, производство активных форм кислорода и митохондриальную динамику (Bindu et al., 2016). Было сообщено, что докс снижает уровни SIRT3, а также митохондриальные белки слияния митофузин-1 (MFN1) и митохондриальный динамин, такой как GTPase (OPA1), и увеличивает повреждение митохондриальной ДНК (мтДНК) и продукцию активных форм кислорода ( Pillai VB et al., 2017). Сводные отчеты об этих результатах представлены в Таблице 1 и на фиг. 1 А.
Таблица 1 . Влияние доксорубицина на кардиотоксичность: отчеты об исследованиях in vitro.
Модель исследованияПротокол исследования (препарат / доза / длительность)основные результатыТолкованиеRef.Окислительный стрессАпоптозФункция митохондрийИсследование на выживание•
Клетки HL-1
•
Докс / 0,3 мкм / 24ч
-
↓pAMPK
-
↔OCR
-
↓жизнеспособность клеток
-
↑P-STAT1
-
↑p-AKT
-
↑p-ERK
-
↑p-38
•
Докс-индуцированная кардиомиопатия, включающая IFNy-перепрограммирование сердечного метаболизма.
Dox/1μm / 24h+
IFNy / 20ng / ml / 24h
-
↓pAMPK
-
↓OCR
-
↓↓жизнеспособность клеток
-
↑ ↑p-stat1
-
↑ ↑p-AKT
-
↑ ↑p-ERK
-
↑ ↑P-38
•
Клетки H9c2
•
Dox / 1μm / 24h
-
↑Активные формы кислорода
-
↑СРЕДНЕСРОЧНАЯ ОЦЕНКА
-
↑Апоптоз
-
↓СПС
-
↓п-АМПК
-
↓PGC1a
-
↓NRF1
-
↓UCP2
-
↓TFAM
-
↓жизнеспособность клеток
•
Докс-индуцированная кардиотоксичность проявляется повышенным оксидативным стрессом, апоптозом и митохондриальной дисфункцией.
Кардиомиоциты мыши, сердечные фибробласты (PAR-1 +/+ vs PAR-1 -/-)
•
Dox / 1μm / 24h
•
PAR -1 + / +
-
Species активные формы кислорода
-
↑супероксид
•
Пар-1-/-
-
Species активные формы кислорода
-
↓супероксид
•
PAR -1 + / +
-
CLE расщепление PARP
•
Пар-1-/-
-
CLE расщепление PARP
•
PAR-1+/+ (CMs)
-
↑деполяризация
•
PAR-1-/- (CMs)
-
↓деполяризация
•
PAR-1+/+
-
↓жизнеспособность клеток
•
Пар-1-/-
-
↑жизнеспособность клеток
•
Dox-индуцированная кардиотоксичность через PAR-1.
Dox/1μm / 24h+
Пар-1/150 мкм / 24ч
•
PAR -1 + / +
-
Species активные формы кислорода
-
↑↑супероксид
•
Пар -1-/-
-
Species активные формы кислорода
-
↑супероксид
•
PAR -1 + / +
-
CLE расщепление PARP
* PAR -1 -/- - CLE расщепление PARP
•
PAR -1 + / + (CMs)
-
↑↑деполяризация
•
PAR -1 - / - (CMs)
-
↑деполяризация
•
PAR -1 + / +
-
↓↓жизнеспособность клеток
* PAR-1 -/- - viability жизнеспособность клеток
•
Первичные кардиомиоциты
•
Dox / 2μm / 24h
•
Dox / 5μm / 24h
-
Species активные формы кислорода
-
↑8-оксо-ДГ
-
↑деполяризация
-
damage повреждение мтДНК
-
↓SIRT3
-
MF РНБ1
-
↓OPA1
-
↓%трубчатых митохондрий
-
↑гибель клеток
•
Докс-индуцированная кардиотоксичность путем уменьшения
Уровни SIRT3.
CMs: кардиомиоциты; Dox: доксорубицин; IFNyInterferon gamma; PGC1a: Пероксисомный пролифератор-активированный рецептор коактиватор 1α; OCR: скорость потребления кислорода; TFAM: митохондриальный фактор транскрипции A; MDA: Малоновый Диальдегид; NRF1: ядерный дыхательный фактор 1; PAR-1: протеаза-активированный рецептор; UCP2: расцепляющий белок 2; 8-oxo-dG: 8-Oxo-2'-дезоксигуанозин.