Если вы сдали тест полногеномного секвенирования (WGS) и получили исходные данные, то наверняка задались вопросом: что с ними делать дальше? Большинство генеалогических сервисов - FTDNA, GEDmatch, YFull и другие - не принимают сырые форматы напрямую, и здесь на помощь приходит WGS Extract - бесплатный инструмент который превращает ваши данные в файлы пригодные для загрузки в любой сервис.
В зависимости от провайдера исходные данные могут отличаться. tellmeGen выдаёт файлы в формате FASTQ. Dante Labs предоставляет как FASTQ, так и уже готовый BAM файл выровненный на референс hs37d5 - в этом случае шаг выравнивания можно пропустить и начать работу сразу с загрузки BAM в программу.
В этой статье я подробно разберу весь процесс работы с WGS Extract: от первого запуска и выравнивания до получения готовых файлов.
Что такое WGS Extract и как его установить
WGS Extract - бесплатная десктопная программа для обработки данных полногеномного секвенирования. Она берёт на себя всю биоинформатическую работу: скачивает референсные геномы, запускает выравниватель BWA-MEM, индексирует файлы - и всё это через простой графический интерфейс без необходимости работать в командной строке.
Скачать программу можно на официальном сайте wgsextract.github.io. Там же находится документация и актуальные релизы. Текущая стабильная версия - Beta v4.
На момент написания статьи вышла новая версия WGS Extract v6 Alpha.
Системные требования
Программа работает на Windows, macOS и Linux. Особых требований к процессору нет - чем мощнее машина, тем быстрее пройдёт выравнивание, но принципиальных ограничений нет.
Гораздо важнее свободное место на диске. С учётом исходных FASTQ файлов, референсных геномов и двух BAM файлов потребуется порядка 300 – 400 ГБ. При этом настоятельно рекомендую использовать SSD, а не HDD - выравнивание на жёстком диске будет значительно медленнее из-за низкой скорости записи.
Скачивание и установка
После скачивания архива с сайта распакуйте его в удобное место и запустите установочный скрипт для вашей операционной системы:
- macOS - запустите файл Install_macos.command и разрешите установку в системных настройках
- Windows - запустите файл Install_windows.bat от имени администратора
- Linux - выполните в терминале bash Install_linux.sh
Скрипт автоматически установит все необходимые зависимости: samtools, BWA-MEM и другие биоинформатические утилиты. После завершения запустите программу через файл:
- WGSExtract.command на macOS
- WGSExtract.sh на Linux
- WGSExtract.bat на Windows
Первоначальная настройка
Требования к диску
Прежде чем запускать программу, важно разобраться с местом на диске - это один из ключевых практических моментов.
В процессе работы WGS Extract создаёт несколько крупных файлов. Вот примерный расчёт необходимого пространства:
- Исходные FASTQ (R1 + R2) - ~100–110 ГБ
- Референсный геном hs37d5 - ~3 ГБ
- Референсный геном T2T - ~4 ГБ
- BAM файл hs37d5 - ~130–150 ГБ
- BAM файл T2T - ~130–150 ГБ
- Итого - ~370–420 ГБ
Объём BAM файлов зависит от реального покрытия ваших данных. Если провайдер заявляет 30x, фактическое покрытие нередко оказывается выше - в таком случае BAM файлы будут крупнее.
Не менее важна скорость диска. Выравнивание - это непрерывный интенсивный процесс чтения и записи, и HDD справится с ним значительно медленнее SSD. На практике разница может составлять в два-три раза по итоговому времени обработки. Оптимальный вариант - внешний NVMe SSD объёмом от 500 ГБ, подключённый через USB-C или Thunderbolt.
Настройка Output Directory
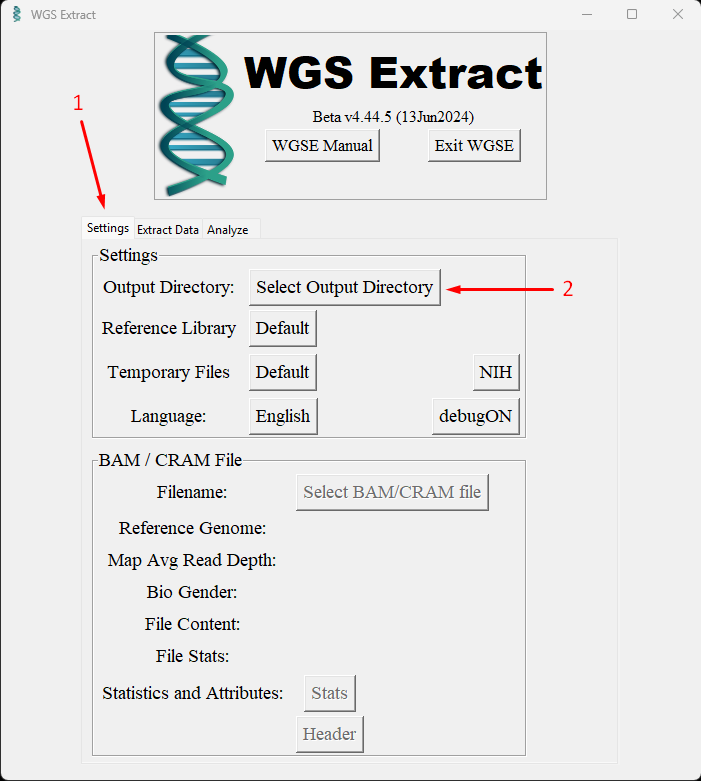
Первое что нужно сделать после запуска программы - задать папку для выходных файлов. Для этого откройте вкладку Settings и нажмите кнопку Output Directory. Укажите папку на диске с достаточным свободным местом.
Туда же рекомендую заранее скопировать и исходные FASTQ файлы - это ускорит работу, так как программа будет читать и писать с одного диска.
Важно: никогда не указывайте в качестве Output Directory папку внутри директории установки самой программы - это может привести к конфликтам при обновлении.
Выравнивание FASTQ в BAM
Выравнивание - это основной и самый длительный этап работы. Программа берёт исходные FASTQ файлы и «укладывает» каждый прочитанный фрагмент ДНК на соответствующее место референсного генома, создавая BAM файл - отсортированную карту вашего генома.
Для разных сервисов нужны BAM файлы, выровненные на разные референсные геномы. Поэтому выравнивание придётся запустить дважды:
- hs37d5 - для получения аутосомных файлов и файла митохондриальной ДНК
- T2T (CHM13 v2.0) - для получения файлов Y-хромосомы и митохондриальной ДНК
Если у вас данные от Dante Labs: вы уже получили готовый BAM файл, выровненный на hs37d5. Подраздел - "Выравнивание на hs37d5" можно пропустить и сразу перейти к следующему разделу. Для YFull потребуется выравнивание на T2T - следуйте подразделу - "Выравнивание на T2T", только вместо FASTQ укажите ваш BAM файл.
Выравнивание на hs37d5
Откройте вкладку Analyze и нажмите кнопку Align. В открывшемся окне выберите ваши FASTQ файлы - оба: R1 и R2. Затем в списке референсных геномов выберите hs37d5 (86 SNs).
Если вы используете этот референсный геном впервые, программа автоматически скачает его - около 3 ГБ. После загрузки задайте имя выходного BAM файла и нажмите подтверждение.
Начнётся процесс выравнивания. Его продолжительность зависит от мощности процессора и скорости диска - от нескольких часов до нескольких суток. По завершении обязательно перейдите к подразделу - "Проверка результатов через Stats".
Выравнивание на T2T
Процесс полностью аналогичен подразделу - "Выравнивание на hs37d5". Откройте вкладку Analyze, нажмите Align, выберите те же FASTQ файлы R1 и R2, но на этот раз укажите референсный геном T2T CHM13 v2.0 (25 SNs). Задайте новое имя для выходного BAM файла и нажмите подтверждение.
Выравнивание на T2T занимает заметно больше времени, чем на hs37d5 - от половины суток до 7 дней в зависимости от мощности вашего компьютера. По завершении обязательно перейдите к подразделу - "Проверка результатов через Stats".
Проверка результатов через Stats
После каждого выравнивания необходимо запустить Stats вручную - без этого шага все остальные кнопки программы останутся заблокированными. Перейдите на вкладку Settings, найдите раздел BAM/CRAM File и нажмите кнопку Stats. Для проиндексированного BAM файла результаты появятся за несколько секунд.
Программа отобразит сводную таблицу с ключевыми показателями. Вот на что обращать внимание:
- Average Read Depth MAPPED - средняя глубина покрытия: сколько раз каждая позиция генома была прочитана. Для 30x теста нормальное значение - от 28x и выше. Значения ниже 20x могут свидетельствовать о проблемах при секвенировании или выравнивании
- Gigabases MAPPED - объём данных, успешно выровненных на референс. Должно составлять около 95% от RAW Gigabases
- Gigabases RAW - общий объём данных в файле (выровненные + невыровненные чтения). Для 30x теста ожидается 90 ГБ и выше
- Read Segs MAPPED - количество индивидуальных ДНК фрагментов, выровненных на референс. Для 30x WGS типичные значения - от 600 до 900 миллионов
- Reads MAPPED (%) - процент ридов, успешно выровненных на референсный геном. Хороший результат - 90% и выше. Значительно более низкий показатель может указывать на несоответствие референса или проблему с исходными FASTQ файлами
- Reference Model - референсный геном, использованный при выравнивании. Убедитесь, что значение соответствует ожидаемому: hs37d5 или T2T CHM13 v2.0
- File Content - содержимое файла. Должно показывать Whole Genome со всеми хромосомами (Auto, X, Y, Mito) . Если отображается только часть хромосом - что-то пошло не так при выравнивании
Если все показатели в норме - выравнивание прошло успешно и можно переходить к следующему разделу.
Получение аутосомных файлов
Аутосомные файлы имитируют результат коммерческого SNP-теста на микрочипе - только с более полным покрытием и меньшим числом пропущенных позиций. Именно эти файлы принимают генеалогические сервисы для поиска родственников и анализа происхождения.
Для этого шага вам понадобится BAM файл, выровненный на hs37d5 - либо созданный в подразделе - "Выравнивание на hs37d5", либо полученный напрямую от провайдера (например, Dante Labs). Убедитесь, что он загружен в программу и Stats был запущен.
Перейдите на вкладку Extract Data. В верхней части вкладки найдите раздел Microarray Test и нажмите большую кнопку "microarray Raw".
Откроется окно выбора форматов. Программа предложит четыре рекомендуемых варианта - нам нужны только два: Combined file of ALL SNPs и v3 + v5. Выберите их и нажмите "Generate".
Первая генерация занимает около 1 часа - программа выполняет полный пайлап и вызов вариантов. Появится окно ожидания - не закрывайте программу до его завершения. Все последующие запуски для того же BAM файла займут менее 2 минут, так как программа использует созданный CombinedKit как кэш.
Важно: не удаляйте файл CombinedKit из Output Directory. Он служит отправной точкой для быстрой генерации любых дополнительных форматов.
Получение файлов Y-хромосомы и мтДНК
Сервисы анализа Y-хромосомы и митохондриальной ДНК не принимают полный BAM файл целиком - им нужна только соответствующая часть генома. WGS Extract позволяет извлечь её в виде компактного файла весом 300–800 МБ вместо исходных 130–150 ГБ.
Вам потребуются два BAM файла, созданных ранее: выровненный на hs37d5 и выровненный на T2T CHM13 v2.0. Работа с файлами ведётся поочерёдно. Сначала загрузите BAM на hs37d5, запустите Stats и выполните нужные операции. Затем загрузите BAM на T2T, снова запустите Stats и повторите.
Для BAM файла, выровненного на hs37d5, перейдите на вкладку Extract Data и найдите раздел Mitochondrial DNA. Нажмите кнопку "MT-only FASTA". Программа извлечёт из полного BAM только чтения митохондриальной ДНК и сохранит ее в отдельный файл - в Output Directory появится файл с суффиксом _mtdna и расширением .fasta.
Для BAM файла на T2T CHM13 v2.0 перейдите на вкладку Extract Data, найдите раздел Y Chromosome и последовательно нажмите две кнопки:
- "Y and MT BAM" - извлекает чтения Y-хромосомы и мтДНК, файл с суффиксом _chrYM
- "Y-only BAM" - извлекает только чтения Y-хромосомы, файл с суффиксом _chrY
Процесс занимает несколько минут.
Для женщин: Y-хромосома в геноме отсутствует, поэтому раздел Y-DNA не актуален. Для анализа митохондриальной ДНК можно использовать кнопку "MT-only FASTA" в том же разделе.
Заключение
Если вы прошли все предыдущие шаги, у вас теперь есть полный набор файлов для загрузки в генеалогические сервисы. Подведём итог: что было сделано и куда что загружать.
Из BAM файла на hs37d5 вы получили аутосомные файлы - они используются для поиска родственников и анализа этнического происхождения и мтДНК - для анализа материнской линии и определения гаплогруппы. Из BAM файла на T2T вы получили файлы Y-хромосомы и мтДНК - они используется для анализа отцовской и материнской линий и определения гаплогруппы.
- CombinedKit - GEDmatch
- 23andMe v35 - FTDNA, Familio и другие похожие сервисы
- MT-only FASTA и Y-only BAM - YFull¹
- Y and MT BAM - TheYtree
- FASTQ - DNAChron и Genotek²
¹ Файл BAM загружается через облачное хранилище, например Dropbox - YFull скачивает его по ссылке.
² Для загрузки в Genotek напишите в службу поддержки и предоставьте ссылку на файлы в облаке, например Облако Mail.
Исходные FASTQ файлы - самый ценный долгосрочный актив. Из них можно в любой момент повторить выравнивание на любой референсный геном, в том числе на те, которые появятся в будущем. Никогда не удаляйте оригинальные FASTQ.
Дополнительно проверить качество BAM файлов можно на сайте bam.iobio.io - бесплатный онлайн-инструмент, который не требует установки и работает прямо в браузере. Он покажет покрытие по каждой хромосоме, процент выровненных ридов, уровень дублирования и распределение качества чтений - всё в виде наглядных графиков и за считанные секунды.