
Тут на днях коллега (Ирина Манто) опубликовала бланк иммунограммы пациента 4 лет с язвенным колитом, и я подумала, а почему бы не написать на эту тему, ибо колиты любого проявления– это не редко первый и единственный симптом какого-нибудь дефекта иммунной системы, а вовсе не "просто обычное ВЗК".
Почему это крайне важно? потому что тактика лечения будет частенько совершенно другая.
Есть такое понятие у гастро врачей- very early onset IBD, это ВЗК очень раннего начала. Обычно ВЗК, когда это проблема жкт, полигенная (множество генов, предрасположенность и риски)– это старт в подростковом и взрослом возрасте. VEO-ВЗК это подгруппа пациентов со стартом ДО 6 лет. Среди этих самых VEO-ВЗК детей (то есть внутри группы <6 лет) примерно 15–20% имеют выявляемый моногенный дефект, то есть конкретную мутацию в одном гене, связанную с иммунной регуляцией или эпителиальным барьером. Остальные ~80% не имеют на сегодня найденной одиночной мутации и по поведению ближе к «обычному» ВЗК. Это (моногенные ВЗК) также составляет примерно 6-11% пациентов из всей группы "детское взк".
Вообще есть целый спектр первичных иммунных дефицитов, которые могут проявляться ВЗК-подобными симптомами/клиникой, я сейчас обо всем взк в целом, а не только VEO-ВЗК. Примерно у 20% генетических дефектов, лежащих в основе первичных иммунных дефицитов, может развиваться воспаление кишечника (то самое ВЗК-подобное). Именно заболевания, связанные с иммунной дисрегуляцией, чаще всего проявляются фенотипом, похожим ВЗК-подобный фенотип: до 40% различных генетических дефектов в этой группе дают такую клиническую картину (видно на картинке).
На сегодня описано более 60 моногенных заболеваний, которые проявляются кишечным воспалением, посмотрите на картинку:
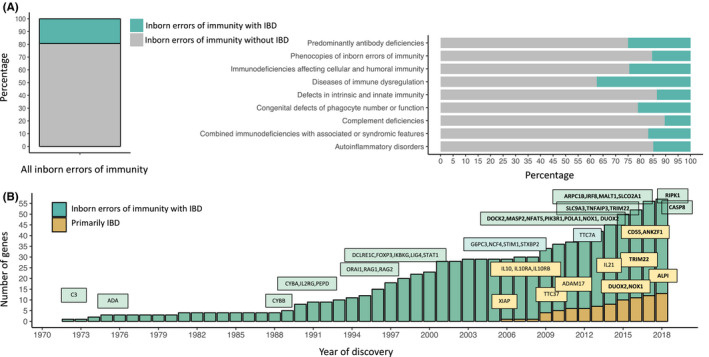
Так вот возвращаясь к VEO-ВЗК, какие моногенные проблемы могут его вызывать? Тут есть споры с терминологией. Ибо ВЗК- это конкретные 2 нозологии, язвенный колит или болезнь крона, с четкими характеристиками, в том числе и гиcтологическими. Есть много, как я выше пишу, иммунных дефектов, где клинически пациента сопровождает хронический колит, хоторый очень похож на ВЗК, но не ВЗК в полном смысле этого определения. В итоге тут все смешано, где то прям истинное ВЗК развивается, где то взк-подобные фенотипы колита.
Сначала перечислим самое "ходовое", что можно, тк такого цельного списка именно кишечных проблем при ПИД я нигде не нашла, все везде частями:
- Дефекты сигнального пути IL-10 (мутации IL-10, IL-10RA, IL-10RB)
- Иммунная дисрегуляция
–FOXP3 мутация, Иммунодисрегуляция–полиэндокринопатия, сцепленная с Х-хромосомой (IPEX-синдром)
–CTLA4 (гаплонедостаточность CTLA4) → лимфопролиферация, цитопении, энтероколит
–LRBA (LRBA-дефицит) (фенотип очень похож на CTLA4-недостаточность, хроническая диарея/колит, цитопении, спленомегалия.)
–STAT3 gain-of-function, STAT1 gain-of-function (системная аутоиммунность (цитопении, эндокринопатии) + хронический колит.)
–IL2RA, STAT5B
–PIK3CD / PIK3R1 (APDS-1 / APDS-2) (лимфопролиферация, синусолёгочные инфекции, энтероколит. Эти синдромы относят к первичным иммунорегуляторным расстройствам).
–XIAP (BIRC4)– Х-сцеплённый лимфопролиферативный синдром типа 2 (Тяжёлый гранулематозный Кроноподобный колит с перианальными поражениями, плюс риск HLH на фоне EBV. Часто требует трансплантации.)
- Дефекты Т- и В-клеток
–Синдром Вискотта — Олдрича
– Тяжёлый комбинированный иммунодефицит (SCID: IL2RG, ADA, DCLRE1C/Artemis, ZAP70 и др + RAG1 / RAG2 (leaky SCID / синдром Оммена)
– Общий вариабельный иммунодефицит (CVID) (у части пациентов бывает хронический гранулематозный или лимфоцитарный колит по типу ВЗК.
– DOCK8 дефицит (тяжёлые вирусные инфекции кожи, высокий IgE, аллергия, энтеропатия с колитом) — часто относят к комбинированной иммунной недостаточности с аллергией и энтеропатией. (Эти случаи описаны как IBD-like у детей.)
- Дефекты фагоцитоза
–Хроническая гранулематозная болезнь (гены CYBB, CYBA, NCF1, NCF2, NCF4)
– ITGB2 (CD18) → дефицит адгезии лейкоцитов типа I (LAD-1)
– G6PC3 (тяжёлая врождённая нейтропения тип G6PC3) и SLC37A4 (болезнь накопления гликогена Ib с нейтропенией)
- Гипервоспалительные и аутовоспалительные заболевания
–Х-сцеплённый лимфопролиферативный синдром 2 типа (XIAP-дефицит)
– Синдром Хермански — Пудлака (Hermansky–Pudlak)
– NLRC4 gain-of-function
– MEFV (Семейная средиземноморская лихорадка) и другие периодические лихорадки
- Нарушение эпителиального барьера
– Дефицит ключевого модулятора NF-κB (NEMO, синдром дефицита обязательного модулятора NF-κB)
– TTC7A
– EPCAM – врождённая «tufting enteropathy»
– FERMT1 (Kindlin-1) → синдром Киндлера
– дефицит ADAM17
Этих мутаций, ВЗК-подобных, где будет поражен жкт элемент, в виде колитов разной степени безобразности, очень много, более 60 штук на сегодня (вот тут табличка чуть больше). Среди всего этого списка есть группа мутаций, которая выглядит именно как ВЗК и является моногенным ВЗК. Ассоциация КРона и ЯК приводят примерно такой же список (чуть менее обширный), как самые "ходовые" мутации при VEO-IBD. НО тут нужно маленькое пояснение:
Для ассоциации Крона и ЯК VEO-IBD = «IBD, начавшееся до 6 лет». И практически всякий ребёнок <6 лет с хроническим воспалительным поражением кишечника, с кровью, свищами, задержкой роста, перианальной болезнью и т.д., попадает под этот зонтик на этапе первичной оценки.
Они сознательно НЕ проводят в этом тексте строгой границы между: истинным классическим ВЗК (болезнь Крона / язвенный колит по морфологии) и тяжёлой энтеропатией / колитом в рамках первичного иммунодефицита.
Это видно по тому, что в их «патологические находки совместимые с VEO-IBD» они включают, кроме гранулём и свищей, ещё и вещи вроде виллезной атрофии, эозинофильной инфильтрации, апоптоза эпителия, повышенных интраэпителиальных лимфоцитов и признаков врождённых энтеропатий. Это не классическая морфология болезни Крона или язвенного колита подростка; это больше картина IPEX, врождённых энтеропатий барьера и тяжёлой иммунной дизрегуляции у младенца. Это нужно понимать, ВЗК и ВЗК-подобные.
В общем и целом все это можно разделить на уровни (это чисто мое прочтение, в литературе все в кучу в этом отношении):
Первый уровень– ситуации, где кишечник у ребёнка выглядит практически как типичный тяжёлый Крон/язвенный колит (по эндоскопии и гистологии), просто это случилось чудовищно рано, а причиной оказалась одна конкретная мутация. Это то, что педиатр-гастроэнтеролог исторически называет «monogenic IBD», и это же чаще всего встречается у малышей с VEO-IBD. Это действительно ядро:
- Путь IL-10: IL10, IL10RA, IL10RB – агрессивный кроноподобный свищевой, гранулематозный колит у младенцев, перианальная деструкция, резистентность к стандартной терапии. Решение тут- трансплантация ТГСК.
- Дефекты NADPH-оксидазы (хроническая гранулематозная болезнь): CYBB, CYBA, NCF1, NCF2, NCF4 – трансмуральный гранулематозный колит со свищами, стриктурами, перианальными абсцессами, то есть классический «очень злой Крон», просто в детском возрасте. (решение опять же трансплантация)
- Дефицит XIAP (BIRC4)– тяжёлый свищевой перианальный кроноподобный колит, риск HLH, часто показание к трансплантации ТГСК.
Второй уровень– Иммунная дисрегуляция, дающая морфологию, похожую на язвенный колит. Здесь кишечник гистологически у многих детей действительно выглядит как хронический активный колит с архитектурной перестройкой крипт, то есть почти как язвенный колит. Но у ребёнка, кроме кишечника, есть системная иммунная проблема (аутоиммунные цитопении, спленомегалия, лимфопролиферация и т.д.).
- CTLA4 (гаплонедостаточность CTLA4)
- LRBA (LRBA-дефицит)
У этих детей бывает очень «настоящий», упорный UC-подобный колит, но при этом видно, что вся иммунная система «разболтана»: лимфопролиферация, цитопении, аутоиммунные проявления вне кишечника. Поэтому формально это относят к «моногенному IBD», но по сути это уже «IBD-подобный колит при иммунной дизрегуляции», а не изолированный Крон/ЯК. Тем не менее CTLA4 и LRBA в современных когортах раннего ВЗК входят в наиболее часто выявляемые гены.
Третий уровень– "все остальное", другие известные моногенные причины раннего ВЗК/ВЗК-подобного колита. Тут идёт длинный «хвост» более редких причин, которые в литературе после 2020 года описываются как моногенные формы раннего ВЗК или тяжёлый IBD-like колит.
- NFKB2, PIK3CD / PIK3R1 (APDS), STAT3 gain-of-function, STAT1 gain-of-function, IKZF2 – иммунная дизрегуляция, хронический тяжёлый колит + системная аутоиммунность/лимфопролиферация.
- TTC7A – тяжелейшее разрушение кишечного барьера и колит у новорождённых с множественными кишечными атрезиями; иногда это называют VEO-ВЗК, иногда отдельно как «катастрофа барьера», потому что морфология не всегда классический ЯК/Крон.
- NOD2, DUOX2, NOX1, TRAF3, NLRP6 / NLRP12 / NLRP2, TRIM22 – дефекты врождённого иммунитета слизистой и контроля микробиоты; описаны у детей с ранним фенотипом ВЗК.
- DOCK8 (комбинированный иммунодефицит с вирусными инфекциями кожи и слизистых) – тяжёлый хронический колит, порой трактуемый как ВЗК у ребёнка, но это ВЗК-like.
- SLC37A4 (болезнь накопления гликогена Ib с нейтропенией), CYBB-гипоморфные варианты, ITGB2 (LAD-1) – Кроноподобный трансмуральный колит со свищами и перианальными язвами у маленьких детей.
- MYO5B, SLC26A3, FERMT1 (Kindlin-1) – тяжёлая ранняя энтеропатия с воспалением толстого кишечника, иногда описываемая клинически как ЯК-подобный колит у грудничка, но патологоанатом видит нарушение полярности/адгезии эпителия, а не классический подростковый ЯК.
И это далеко не весь перечень, как я выше писала, на сегодня суммарно описано уже более 60 отдельных моногенных состояний, которые могут дать у ребёнка фенотип «как ВЗК» (Крон/ЯК-подобный), особенно если дебют до 6 лет.
Делать то что?
Но это все прекрасно, а делать то врачу что? как поймать такое и что с ним делать, ведь каждого пациента не отправишь сразу на генетику? Ассоциация крона и ЯК дает вполне себе алгоритм, я просто его приведу тут, оригинал опять же по ссылочке можете прочесть.
Ребенок с придет к вам на приме с ворохом симптомов, что абсолютно НЕ специфичны ВЗК и могут быть при куче других патологий:
- кровь и/или слизь в стуле,
- упорная диарея,
- частая рвота,
- не набирает вес / не растёт,
- перианальные изменения (свищи, кожные метки, глубокие трещины),
- эпизодические лихорадки,
- воспалительные суставные жалобы (артрит, артралгии),
- внекишечные проявления типа фолликулита, увеита.
Руководство подчёркивает, что при таком наборе жалоб у ребёнка до 6 лет врач ОБЯЗАН сначала вычистить более банальные причины. Это и есть дифференциальная диагностика. Она включает:
- Инфекции кишечника: Salmonella, Shigella, Campylobacter, патогенные эшерихии (EHEC/EPEC), Giardia, Cryptosporidium, а также Clostridioides difficile (у детей старше 12 мес; у младенцев токсин C. diff часто неинтерпретируем).
- Пищевая аллергия/аллергический колит, непереносимость белков коровьего молока.
- Целиакия.
- Дефицит питания/калорий, другие метаболические причины плохого роста.
- Другие воспалительные или аутоиммунные заболевания (включая тиреоидную патологию).
- Первичные иммунодефициты и наследственные синдромы иммунной дисрегуляции.
То есть уже на уровне дифференциала врачи должны думать не только «это Крон», но и «а не врождённая иммунная ошибка ли это», особенно если есть тяжёлые инфекции, семейный анамнез иммунных проблем, кровное родство родителей (редкость для нас, но все же), множественные выкидыши в семье, признаки лимфопролиферации (спленомегалия, лимфаденопатия).
Ну и документ нам дает пошаговый алгоритм, что делать, какие тесты проводить, если перед вами ребенок до 6 лет с симптомами, похожими на ВЗК:
Первичная настороженность
- Ребёнок <6 лет.
- Есть хронические симптомы (кровавый стул, диарея, перианальная болезнь, плохой рост, повторные лихорадки и т.д.).
- На этом этапе тщательно собирают анамнез (включая инфекции и аутоиммунные проявления вне кишечника) и смотрят физикально на кожу, рот, перианальную область, лимфоузлы, печень/селезёнку, задержку роста. Всё это нужно, чтобы уловить «подсказки» моногенного дефекта.
Базовое обследование (первый ряд)
Это то, что делают практически всем, до всякой экзотики. Цель — отсечь банальные и частые причины:
- Общий анализ крови (анемия, тромбоцитоз, лейкоцитоз/лейкопения).
- Биохимия (печёночные ферменты, альбумин и пр.).
- Маркёры воспаления (СОЭ, С-реактивный белок).
- Кальпротектин или лактоферрин в стуле как кишечные маркёры воспаления.
- Скрытая кровь в кале.
- Копрологические/молекулярные тесты на бактериальные, паразитарные и токсин-продуцирующие возбудители (см. список выше).
- Исключение целиакии и аллергического колита, проверка щитовидки, при необходимости нутритивного дефицита.
- Эндоскопия: эзофагогастродуоденоскопия + колоноскопия с попыткой зайти в терминальный отдел подвздошной кишки.
- Гистология с прицельным описанием: ищут признаки, согласующиеся с ВЗК или врождённой энтеропатией (эозинофильные инфильтраты; виллезная атрофия; апоптоз эпителия; повышенные интраэпителиальные лимфоциты; гранулёмы; макрофаги с пигментом и т.д.).
- Визуализация тонкой кишки (узи тонкой кишки, МР-энтерография, КТ-энтерография) если нужно оценить протяжённость, утолщение стенки, свищи.
Если на этом этапе нашлось простое объяснение (инфекция, аллергический колит на белок коровьего молока и т.д.) — это не VEO-IBD.
Если же после этой первой линии остаются признаки настоящего воспалительного заболевания кишечника (особенно если есть тяжёлое поражение толстой кишки, перианальные свищи, гранулёмы, системные проявления), — идём дальше.
Второй ряд» (second-tier testing)
Это более углублённые исследования иммунной системы, которые помогают понять, не скрывается ли под видом ВЗК врождённая ошибка иммунитета / моногенное заболевание. Руководство перечисляет:
- Уровни иммуноглобулинов (IgA, IgM, IgG, IgE).
- Поствакцинальные титры антител (и параллельно, естественно, уточняют календарь прививок).
- Иммунофенотипирование (T/B/NK субпопуляции).
- Тест окислительного взрыва нейтрофилов (для исключения хронической гранулематозной болезни).
- Тестирование на ВИЧ.
- Скрининг на туберкулёз (Quantiferon / T-Spot).
Смысл шага: не пропустить тяжёлый первичный иммунодефицит, моногенную форму ВЗК (IL-10 дефект, XIAP, CGD и т.д.), состояния, в которых стандартная терапия ВЗК будет либо бесполезна, либо опасна.
Генетика
Если второй ряд даёт повод подозревать моногенную форму, обсуждается генетическое тестирование. Руководство подчёркивает:
Не каждому ребёнку с подозрением на VEO-IBD сразу нужно секвенирование!!!!
Секвенирование ориентируют на тех, кто:
- дебютировал до 2 лет,
- имеет фенотип, характерный для известных моногенных дефектов,
- родственный брак в семье,
- есть яркие отклонения иммунограммы.
ТО есть нужна очень высокая степень подозрений, чтобы отправлять на генетику. Для этого нужно знать клинику этих моногенных дефектов, их иммунологию.
Сегодня у нас есть разные инструменты для генетики и тоже несколько уровней:
- Таргетные панели VEO-IBD (набор известных генов).
- Если панель отрицательна, можно рассмотреть полное секвенирование экзома (WES);
- Если и WES ничего не дал, а клиническое подозрение на моногенный дефект остаётся высоким, обсуждают полное секвенирование генома (WGS).
Решение о генетике, подчёркивают авторы, лучше принимать мультидисциплинарно (гастроэнтеролог + иммунолог + генетик), с до- и послетестовым генетическим консультированием семьи. Это прямо подчёркнуто: семья должна понимать риски, стоимость, страховое покрытие, и то, что иногда исследование идёт в рамках исследовательского протокола.– я это пишу, но конечно для россии это мало актуально, у нас такое редко когда идет в рамках исследовательского протокола, семья сама отстегивает денюжку за тест (это дорого).
Почему так важно выявлять причину VEO-ВЗК?
Потому что если вы этого не сделаете и ограничитесь просто "это ВЗК" лечим по протоколу- вы можете навредить! При некоторых ген дефектах нельзя использовать классическую терапию ВЗК, вы можете ухудшить ситуацию.
Примеры?
- При дефекте IL-10/IL-10R ребёнок должен получить трансплантацию костного мозга как патогенетическое лечение (иначе болезнь остаётся фатальной/неуправляемой). Без диагностики вы лишите этой возможности ребенка и потом будет поздно.
- При XIAP-дефиците ребёнок не только имеет тяжёлый фистулизирующий колит, но и риск смертельного гемофагоцитарного лимфогистиоцитоза на фоне EBV-инфекции; этот риск должен быть известен врачам заранее, чтобы его "поймать".
- При CGD стандартные анти-TNF препараты, которые гастроэнтеролог даёт подросткам с болезнью Крона, считаются опасными (инфекционные осложнения). Это полностью меняет терапевтическую тактику. Они просто напросто запрещены.
Документ отдельно напоминает, что ребёнок с VEO-IBD — это не всегда «кишечник и только кишка». Врачу велят смотреть и документировать:
- анемию, бледность, отставание в росте, «failure to thrive»
- лимфоаденопатию, спленомегалию (признаки лимфопролиферации)
- кожные поражения (фолликулит, экзема, пиодерма гангренозум, узловатая эритема)
- выраженную перианальную деструкцию (глубокие трещины, свищи, «skin tags»)
- артрит/артралгии
- рецидивирующие инфекции
Потому что эти внекишечные признаки служат маяками к конкретной нозологии:
- Экзема + тромбоцитопения наводит на мысль о синдроме Вискотта–Олдрича.
- Тонкие волосы, аномалии зубов → подозрение на дефицит NEMO.
- Инфантильный колит + тяжёлая перианальная болезнь + фолликулит + артрит → подумать об IL-10 сигнальном дефекте.
- Плохой контроль инфекций и гранулёматозное воспаление → подумать о хронической гранулематозной болезни.
- Колит с выраженной перианальной фистулизацией + риск гемофагоцитоза на EBV → подумать о XIAP-дефиците.
Есть еще одна БОЛЬШАЯ особенность. Тут мы рассматриваем крайние случаи полного фарша симптомов, тк бОльшая часть этих ген нарушений тяжела я идает калейдоскоп симптомов, и главное– стандартные подходы к ВЗК как правило не помогают, лечишь, а не проходит. И это сразу наталкивает на мысль о генетике. НО! есть ситуации с гипоморфными вариантами, когда мутации есть, но они "не полные", проявляют себя частично, тк белок немножко, но производиться, рецептор немножко, но сигналит и тд и тп. И тут возмодны ситуации, что ребенок с ранним дебютом ВЗК ответит изначально на стандартную терапию тем же будесонидом, но через полгода даст громадный рецидив, снеся все на своем пути. Такие дети, с ранним дебютом ВЗК должны быть под мониторингом врачебным даже несмотря на "хороший ответ на базовую терапию".
Будьте здоровы.