Статья подготовлена изданием Лаборант - журнал об аналитической химии. Подписывайтесь так же на наш телеграм-канал о научных новостях Клеточная биология.
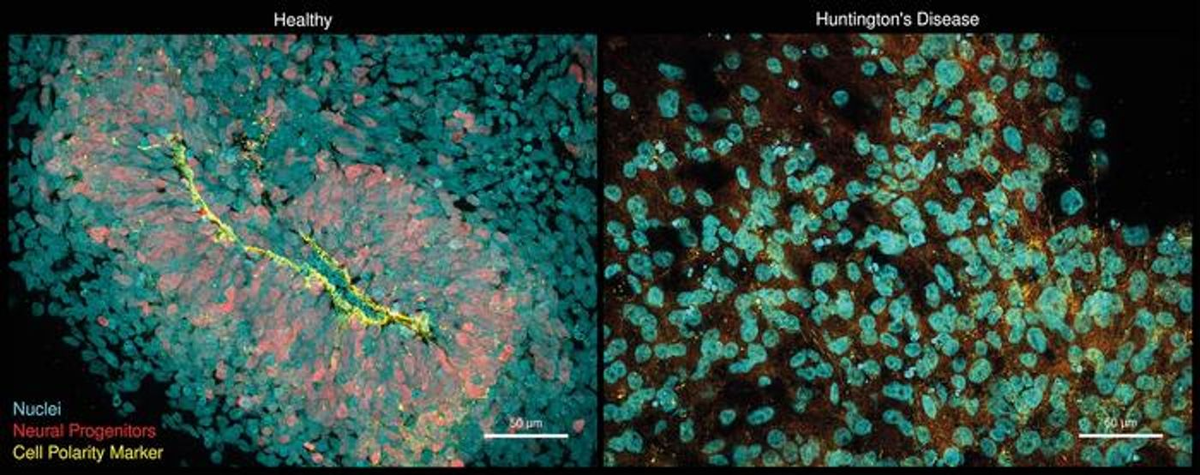
Ученые использовали модель органоидов, чтобы получить новые данные о болезни Хантингтона, смертельном генетическом заболевании, которое вызывает нейродегенерацию, нарушающую движение и поведение. Болезнь вызывается мутациями в гене Huntingtin (HTT). В настоящее время нет эффективных методов лечения этого заболевания. Исследователи обнаружили, что ген под названием CHCHD2 также связан с болезнью Хантингтона; мутации в гене HTT могут снижать экспрессию CHCHD2, что может открыть новые возможности для лечения. Эти результаты были опубликованы в журнале Nature Communications.
Исследователи обращаются к моделям, когда хотят больше узнать о механизмах, лежащих в основе физиологических процессов или заболеваний. Но животные модели могут дать лишь ограниченное понимание человеческих состояний. Одним из относительно недавних изобретений, направленных на преодоление разрывов в исследовательских знаниях, возникающих при использовании животных, являются органоиды. Это упрощенные, миниатюрные модели человеческих органов, созданные путем генетической модификации клеток, чтобы они приняли идентичность типов клеток, специфичных для определенных органов.
В данной работе ученые создали мозговые органоиды, несущие мутации, которые, как известно, вызывают болезнь Хантингтона. Хотя повторения в гене HTT не вызывают заболевание, если их количество меньше 35, большее количество повторений связано с болезнью Хантингтона. Мутации вызывают гибель нейронов в мозге. Как объяснил соавтор исследования доктор Якоб Метцгер из Университета Генриха Гейне в Дюссельдорфе (HHU), с увеличением числа повторов тяжесть заболевания обычно усиливается.
Модель церебрального органоида Хантингтона показала, что экспрессия CHCHD2 была аномально низкой на разных стадиях развития. Снижение уровня CHCHD2 замедляло метаболизм нейронов. CHCHD2 обычно поддерживает здоровье митохондрий и ранее был связан с болезнью Паркинсона. Это первый случай, когда его связывают с болезнью Хантингтона. Исследователи также определили, что при восстановлении функции CHCHD2 метаболизм нейронов возвращается к норме.
"Это было неожиданно," отметила соавтор исследования Селене Ликфетт, аспирантка HHU. "Это в принципе предполагает, что этот ген может быть мишенью для будущих терапий."
Это исследование показало, что мутации в HTT также, по-видимому, нарушают развитие мозга задолго до появления симптомов болезни Хантингтона. Это подчеркивает важность ранней диагностики, добавила Ликфетт.
Органоиды показали, что дефекты возникали в нейрональных предшественниках до появления токсических агрегатов, которые являются характерной чертой болезни Хантингтона. Патология может начинаться у пациентов задолго до того, как она станет обнаруживаемой в клинике.
Болезнь Хантингтона обычно считается прогрессирующей по мере дегенерации зрелых нейронов. Но могут происходить изменения гораздо раньше, и может быть возможно разработать методы лечения для более ранних стадий, предположили исследователи.
"Наши стратегии редактирования генома, в частности удаление региона CAG-повторов в гене Хантингтона, показали большие перспективы в обращении некоторых наблюдаемых дефектов развития. Это предполагает потенциал для генной терапии," сказал соавтор исследования профессор Алессандро Прижоне из HHU.
Увеличение экспрессии CHCHD2 может быть еще одной возможностью, добавил Прижоне, который также отметил, что эти результаты могут иметь значение для нейродегенеративных заболеваний в целом. Дефекты митохондрий могут открыть новые пути для лечения этих заболеваний.
Источники: Центр молекулярной медицины Макса Дельбрюка, Nature Communications
Ведущий научный социальный сетевой сайт и производитель образовательных виртуальных мероприятий и вебинаров.
Источники: Центр молекулярной медицины Макса Дельбрюка, Nature Communications
Статья подготовлена изданием Лаборант - журнал об аналитической химии. Подписывайтесь так же на наш телеграм-канал о научных новостях Клеточная биология.
