Исследователи из Чикагского университета (UChicago) разработали статистический метод, который помогает выявлять гены, вызывающие заболевания.
На январь 2024 года возникновение заболевания у человека часто является совокупным результатом воздействия факторов окружающей среды, несбалансированного питания, плохого психического здоровья и взаимодействия между генами, вызывающими заболевания.
Первые три фактора более или менее подвластны людям, но они никак не могут остановить активность генов, вызывающих заболевания в их организме. Однако если врачи смогут выявить такие гены, это поможет им предотвратить возникновение многих заболеваний или хотя бы ограничить их влияние на общее состояние здоровья пациентов.
В отличие от существующих методик, которые фокусируются на одном гене, новый подход, получивший название causal-Transcriptome-wide Association studies (cTWAS), учитывает множество генов и их вариантов, связанных с заболеванием, что позволяет получать результаты с меньшим количеством ложных срабатываний.
Со слов автора исследования Синь Хэ, если рассматривать гены по одному, то будут ложные срабатывания, но если рассматривать все близлежащие гены и варианты вместе, то вероятность найти причинный ген гораздо выше.
В настоящее время ученые используют genome-wide association studies (GWAS) - метод, который предполагает изучение различий между геномными последовательностями двух групп: в одной группе - пациенты с общим заболеванием, а в другой - нормальные, здоровые люди.
Ученые используют эти различия для выявления генов, ответственных за заболевание. Однако GWAS может выявить только связь между генами и болезнью, но не корреляцию между различными генами, вызывающими заболевание. В блоке может быть много генетических вариантов, которые коррелируют с риском заболевания, но люди не знают, какой из них на самом деле является причинным вариантом. В этом и заключается основная проблема GWAS.
Популярным решением для преодоления таких проблем является использование eQTLs (expression quantitative trait loci) - геномных участков, связанных с изменениями в уровне экспрессии генов. Они выявляют взаимосвязь между различными генами, их вариантами и степенью их вклада в различные генетические проявления. Однако, по словам авторов исследования, когда ученые используют GWAS с eQTLs, это приводит к увеличению числа ложноположительных результатов более чем в 50% случаев.
Именно здесь cTWAS может сыграть огромную роль. Он объединяет данные GWAS и eQLT для точного определения генов, вызывающих заболевания, и их вариантов, и в то же время использует передовые статистические методы для минимизации числа ложноположительных результатов, используя байесовскую модель множественной регрессии.
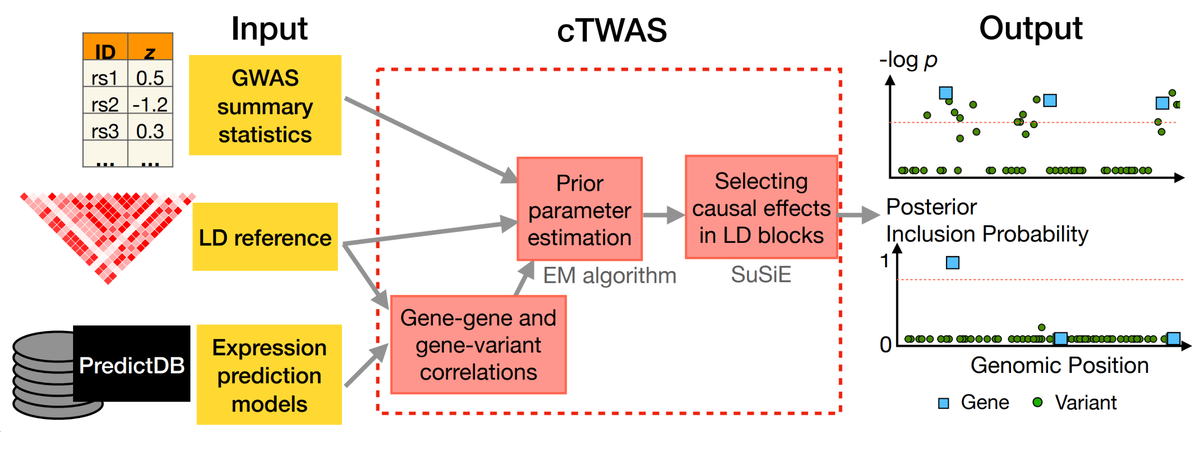
На основе своего метода исследователи разработали программное обеспечение cTWAS, которое можно загрузить со страницы их лаборатории на GitHub. Это программное обеспечение позволит людям проводить анализы, связывающие генетические вариации с фенотипами. Это действительно ключевая проблема, стоящая перед всей областью. Теперь у медицины есть гораздо лучший инструмент для установления таких связей.
Ученые также использовали свой подход для выявления генов, связанных с уровнем холестерина ЛПНП (также называемого плохим холестерином). Они обнаружили 35 предполагаемых причинных генов, связанных с этим заболеванием; более 50% этих генов были обнаружены впервые, что позволяет предположить новые цели и методы лечения ЛПНП.
Ученые планируют продолжить исследование, чтобы усовершенствовать cTWAS и изучить другие возможности его использования.