
Что это такое "неонатальный скрининг "? И зачем его делать, ведь у нас в семье все здоровы?
С 2023 года в работу неонатальных служб Российской Федерации включена программа расширенного скрининга новорожденных. Тестирование новорожденных на наличие наследственных заболеваний вырасло с 5 до 36 нозологий.
ЦЕЛИ ПРОГРАММЫ
- ранняя диагностика наследственных заболеваний;
- оказание адекватной медицинской помощи детям с выявленными заболеваниями;
- сопровождение и реабилитация детей с наследственными заболеваниями;
- снижение инвалидизации пациентов;
- повышение продолжительности и качества жизни детей с выявленными заболеваниями
Взятие крови для неонатального скрининга
Образец крови берут не ранее, чем через 3 часа после кормления в возрасте 24-48 часов жизни у доношенного и 144-168 часов (7 сутки) у недоношенного новорожденного. Поэтому до взятия скрининга новорожденных не кормят 3 часа.
Кровь берут только из пятки. Так же возможно использование венозной крови взятой из пробирки с ЭДТА, если по состоянию здоровья ребёнку показано взятие венозной крови на другие исследования.
Для проведения скрининга берётся 2 тест-бланка с областью для информации о пациенте и пятью кругами - области для нанесения крови.
Капли крови поочерёдно наносят на круги, заполняя 5 кругов на одном бланке и 3 круга на другом.
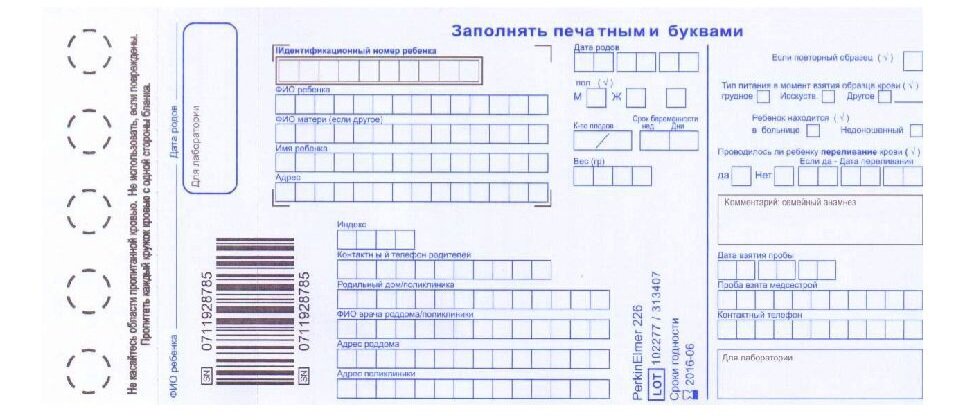
Проведение анализа
Бланки-направления с прикрепленными тест-бланками отправляются в региональную медико-генитическую консультацию (Медико-генетическая консультация располагается на 5-м этаже ГБУ РО «Областной клинический перинатальный центр» г. Рязань), где проводится скрининг на 5 заболеваний. А тест-бланк с 3 пятнами крови отправляется в центр, проходящий исследование не методом ТМС.
36 заболеваний, входящих в неонатальный скрининг
Первые 5 нозологий:
- Фенилкетонурия
- Муковисцидоз
- Адреногенитальный синдром
- Галактоземия
- Врожденный гипотериоз
Новая, включённая в 2023 году в скриниг, 31 нозология
- Спинальна мышечная атрофия (СМА)
- Первичные иммунодефициты
- Дефицит синтеза биоптерина
- Дефицит реактивации биоптерина
- Тирозинемия, тип 1
- Болезнь "кленового сиропа"
- Гомоцистинурия
- Пропионовая ацидемия
- Метилмалоновая ацидемия (6 типов)
- Изовалериановая ацидемия
- Глутаровая ацидемия, 1 тип, 2 тип
- 3-гидрокси-3-метилглутаровая недостаточность
- Первичная карнитиновая недостаточность
- Среднецепочечная ацил-КоА дегидрогеназная недостаточность
- Длинноцепочечная 3-0Н ацил-КоА дегидрогеназная недостаточность
- Очень длинноцепочечная ацил-КоА дегидрогеназная недостаточность
- Недостаточность митохондриального трифункционального белка
- Недостаточность карнитинпальмитоилтрансферазы тип 1, тип 2
- Недостаточность карнитин/ацилкарнитинтранслоказы
- Цитруллинемия тип 1
- Аргиназная недостаточность
- Недостаточность синтеза голокарбоксилаз
- Бета-кетотиолазная недостаточность
- Дефицит биотинидазы
Немного подробнее о данных патологиях
Фенилкетонурия -
это наследственные заболевания, характеризующиеся нарушением метаболизма аминокислоты. Это целая группа заболеваний различной тяжести. С возрастом дети имеют тяжелое поражение нервной системы вплоть до умственной отсталости и эпилепсии. При своевременно назначенном лечении жалобы имеют более легкий характер или отсутствуют.
В России примерно 1 на 7-8 тысяч новорожденных детей, а всего более 200 детей в год, рождается с этим заболеванием.
Муковисцидоз -
генетическое заболевание, при котором нарушен нормальный отток слизи из дыхательных путей, что приводит к застою мокроты и воспалению, характеризуется тем, что в большей степени вовлекаются органы дыхания, поджелудочная железа, печень, желчные пути, желудочно-кишечный тракт, потовые железы. Адекватное своевременное и регулярное лечение, а также реабилитация способствуют улучшению прогноза и качества жизни у большинства пациентов.
В РФ частота муковисцидоза составляет по данным ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова» 1:9000 новорожденных.
Адреногенитальный синдром -
это заболевание, связанное с нарушением работы надпочечников.
Это заболевание, поддающееся лечению. При адекватной терапии и качественном наблюдении за пациентами они ведут нормальный образ жизни, полностью социально адаптируются, получают образование, создают семьи, воспитывают детей.
В РФ распространенность заболевания в отдельных регионах составляет от 1:5000 до 1:12000. Неклассическая форма ВДКН встречается чаще –от 1:500 до 1:1000, а в некоторых изолированных этнических группах, может доходить до 1:50 до 1:100.
Галактоземия -
это одна из наследственных болезней обмена веществ. Это достаточно редкое заболевание. Галактоземия возникает, если у ребенка отсутствует способность расщеплять сахар, содержащийся в молоке.
При отсутствии лечения дети с галактоземией тип 1 погибают на первом году жизни. Если лечение начато рано, то клинические симптомы галактоземии у ребенка не проявятся, и он может расти здоровым, практически не отличаясь от сверстников.
Частота галактоземии в среднем в мире составляет 1: 60 000- 1: 70 000.
Врожденный гипотиреоз —
это одно из наиболее часто встречающихся врожденных заболеваний щитовидной железы у детей, в основе которого лежит полная или частичная недостаточность тиреоидных гормонов, приводящая к задержке развития всех органов и систем организма при отсутствии своевременно начатого лечения. Распространенность в Российской Федерации составляет 1 случай на 3600 новорожденных.
Спинальная мышечная атрофия -
это генетическое, прогрессирующее, иногда смертельное нервно-мышечное заболевание.
Расчетная частота рождения ребенка со СМА 1 на 5184 новорожденных.
Первичный иммунодефицит (ПИД)
представляет собой врожденные нарушения иммунной системы. Заболевание приводит к развитию тяжелых хронических инфекций, а также к аутоиммунному и воспалительному поражению органов и тканей. В большинстве случаев выявить причину развития заболевания не удается. В основе лечения лежит терапия иммуноглобулином человека нормальным. Многое зависит от своевременности назначения терапии (начинать терапию следует сразу после постановки диагноза).
Тирозинемия 1 типа -
это редкое заболевание, при котором организм не усваивает вещество «тирозин», поступающее с пищей. При своевременном начале лечения прогноз, как правило, благоприятный. Диету пациенту необходимо соблюдать в течение всей жизни. После того как диагноз установлен, и поддерживается стабильный уровень тирозина и сукцинилацетона в крови с помощью лекарственной терапии, диеты и специализированных продуктов лечебного питания, ребенок может вести нормальную жизнь и заниматься тем, чем ему хочется, – так же, как и все остальные дети.
Болезнь "кленового сиропа" -
это редкое наследственное заболевание, при котором нарушаются процессы, связанные с расщеплением белка. При отсутствии соответствующей терапии состояние пациента может резко ухудшиться, привести к развитию метаболического криза, необратимым последствиям вплоть до летального исхода.
Гомоцистинурия -
это группа наследственных заболеваний, при которых повышается уровень гомоцистеина в крови. Дети с этим заболеванием не имеют особенностей при рождении. Первыми неспецифическими симптомами могут быть задержка психомоторного развития, умственная отсталость и другие.
Пропионовая ацидемия -
это редкое наследственное заболевание, связанное с дефицитом фермента необходимого для метаболизма ряда аминокислот, а также холестерина, некоторых жирных кислот и свободного пропионата в кишечнике. Наиболее опасными для жизни состояниями, ведущими при отсутствии лечения, к необратимым последствиям вплоть до летального исхода, являются «метаболические кризы».
Нарушение митохондриального бетта-окисления жирных кислот
Клинические проявления: может манифестировать в любом возрасте, проявляется быстро прогрессирующим метаболическим кризом после длительного голодания или физической нагрузки. Тошнота, рвота, часто при нормальном уровне глюкозы в крови. При отсутствии терапии может привести к судорогам, коме и остановке сердца.
Глутаровая ацидурия тип 1 –
наследственное заболевание, связанное с мутациями в гене, который отвечает за работу фермента, необходимого, чтобы происходил правильный метаболизм белков в организме. Проявляется обычно в возрасте от 3 до 36 месяцев. В большинстве случаев первые симптомы заболевания развиваются остро, в виде так называемого энцефалитоподобного или метаболического криза (жизнеугрожающее состояние, проявляющееся в виде изменения уровня сознания, рвоты, судорог). Эффективность лечения во многом зависит от сроков установления правильного диагноза и назначения патогенетической терапии.
Выводы
Практически все заболевания, входящие в неонатальный скрининг, поддаются лечению, если диагноз установлен сразу после рождения. Порой появление жалоб указывает на упущенное время и тяжелое течение патологии, а лечение на этом этапе уже не эффективно. И, поэтому, чем раньше известен диагноз, тем эффективнее лечение и диетотерапия. Ребенок может вести нормальную жизнь и заниматься тем, чем ему хочется, – так же, как и все остальные дети. Адекватное своевременное и регулярное лечение, а также реабилитация способствуют улучшению прогноза и качества жизни у большинства пациентов.
*имеются противопоказания, необходима консультация специалиста