Давайте по порядку.
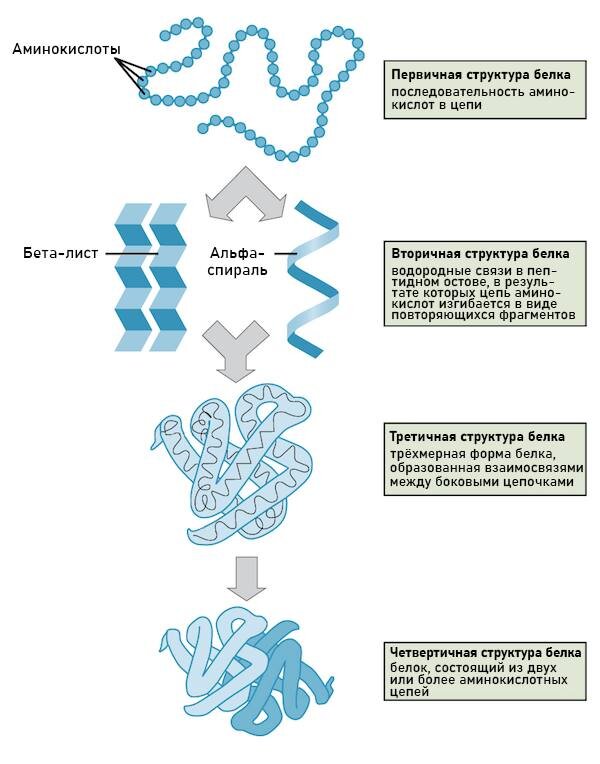
1️⃣Есть ДНК🧬, в ней закодирована первичная структура белковой молекулы (фото 1) — последовательность аминокислот, связанных пептидными связями (C — N) вытянутая в пространстве.
Но в таком состоянии она, во-первых, уязвима, а во-вторых, не функциональна, у неё нет никаких свойств, это просто обычная молекулярная цепь.
2️⃣Вторичная структура - цепь сформировалась и начинает сворачиваться за счет действия внутримолекулярных механизмов основной цепи. На этом этапе она фиксирована водородными связями между кислотной и аминогруппой аминокислот.
🧩Уже здесь появляются различия - какие то белки сворачиваются в спираль, какие то в изменяющуюся в плоскости структуру - бета-складку.
3️⃣Третичная структура - конечная структура для одной белковой молекулы, но не для белка в общем. Здесь молекула сворачивается в ту форму, которая обуславливается взаимодействием не основной цепи аминокислот, как раньше, а боковых - а эти-то взаимодействуют абсолютно по-разному - те же самые водородные связи, ионные взаимодействия, гидрофобные, дисульфидные (фото 2).
🧩На этом этапе сворачивание может остановиться. Чаще всего тут это фибриллы (коллаген например) и глобулы (некоторые ферменты, инсулин и др).
4️⃣Ну и четвертичная - несколько по-разному свернутых цепей, всевозможно связанных.
🔎Итак, пойдем дальше.
Зная последовательность аминокислот мы можем предсказать, как свернется белок - таким образом сотни тысяч человеческих белков уже обнаружены и мы знаем, как они выглядят, грубо говоря, в 3D.
✔️Почему это важно?
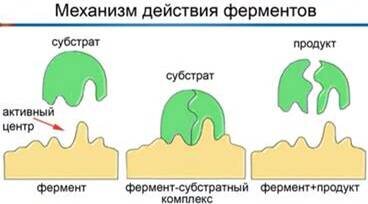
🔓На белках есть в прямом смысле углубления - сайты связывания, в которых, грубо говоря, есть определенная последовательность с определенным зарядом и это углубление является замочной скважиной. (Такие места ещё называются активными центрами, их по несколько у белков.)
🗝Подобрав нужный ключ, мы можем с ними взаимодействовать.
Раньше работало даже без ИИ.
⚙️В программу загружается нужная структура (можно даже не весь белок, а только сайт связывания, с которым нам нужно работать) после чего она пробивает по уже известным в базе белкам, рассчитывая, какой нам нужен субстрат* - то, что подойдёт под известный сайт именно такой формой и таким зарядом, какой нужно.
*scaffold - поправьте, если перевел не так, нормального перевода нет, это по логике чисто.
О чем собственно статья?
☑️Первый способ, который изобрели в прошлом году, немного похож на вышеописанный.
⚙️Мы загружаем в программу нужный сайт связывания и какие-то ключевые особенности строения белка, на который нам нужно воздействовать, а искусственный интеллект, исходя из проанализированных последовательностей сворачивания белков подбирает такую последовательность аминокислот, которая свернётся именно таким образом, чтобы подойти под искомый.
☑️А второй способ уже поинтереснее.
Предположим, нам нужен белок, обладающий определённым свойством. (Например, связывать металл)
⚙️Мы загружаем это в «дано», а в ответ ИИ начинает поэтапно генерировать виртуальный белок из разных последовательностей аминокислот. Разные последовательности в дальнейшем скалыдываются в разные пространственные формы, и тихой сапой подходит к нужному нам варианту.
👨🏻🔬Потом, естественно, в лаборатории можно все эти белковые последовательности создать и свернуть в реальности.
⚠️Конечно, есть недостатки.
Полученный белок может получиться более/менее активным, в результате чего организм может просто не принять его.
Но это все равно хорошо, ведь с полученным результатом можно поработать и в дальнейшем сделать то, что нужно)
✅Результаты:
Были созданы белки, способные:
⁃ связываться с рецепторами раковых клеток
⁃ захватывать металлы
⁃ связывать углекислый газ
⁃ идентифицировать вакцины против респираторно-синцитиальвного вируса
https://www.science.org/content/article/dreaming-new-proteins-ai-churns-out-possible-medicines-and-vaccines собственно статья
https://ru.khanacademy.org/science/biology/macromolecules/proteins-and-amino-acids/a/orders-of-protein-structure тут можно подробнее про структуры белков почитать (картинки оттуда)