Reperfusion injury
Ischemia is characterized by the reduction of blood flow, usually as a result of mechanical obstruction within the arterial system (eg, thrombus). If the flow of blood to the ischemic tissue is restored in a timely manner, those cells that were reversibly injured will typically recover. Sometimes, however, the cells within the damaged tissue will paradoxically die at an accelerated pace through apoptosis or necrosis after resumption of blood flow. This process is termed reperfusion injury, and is thought to occur secondary to one or more of the following mechanisms:
- oxygen free radical generation by parenchymal cells, endothelial cells, and leukocytes;
- severe, irreversible mitochondrial damage described as "mitochondrial permeability transition";
- inflammation, which attracts circulating neutrophils that cause additional injury; and
- activation of the complement pathway, causing cell injury and further inflammation. When the cells within heart, brain, or skeletal muscle are injured, the enzyme creatine kinase leaks across the damaged cell membrane and into circulation (as seen in this patient).
- Cellular swelling arises secondary to changes in ion concentration and the influx of
- water. This state is considered a hallmark of reversible injury, and is not directly associated with the leakage of intracellular proteins such as creatine kinase.
- Glutathione peroxidase actually reduces cellular injury by catalyzing free radical breakdown. The presence of this enzyme is not responsible for the release of creatine kinase.
- Mitochondrial vacuolization reduces the cellular capacity for ATP generation and is associated with irreversible injury. Creatine kinase release is not directly associated with this mitochondrial change, however.
- Nuclear shrinkage (pyknosis), fragmentation, and dissolution characterize irreversible injury of the cell. Creatine kinase release is not directly associated with such nuclear changes, however.
Collagen subtypes
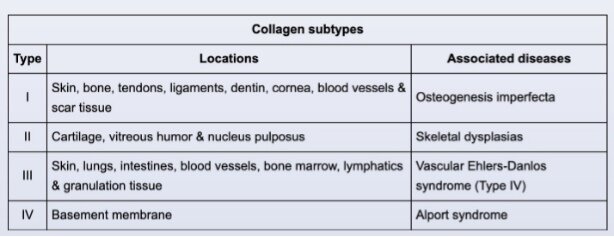
Evolution of a myocardial infarction occurs in several stages. The final stage of the healing process begins approximately 2 weeks after infarction and involves increased deposition of type I collagen. Fibrosis continues until about 2 months after infarction, resulting in a
dense collagenous scar composed primarily of type I collagen. The scar can be identified grossly as a firm, gray-white lesion or microscopically as collagenous tissue replacing myocardium (as in this patient).
In addition to its role in wound healing, type I collagen (the most abundant type) provides strength and support for a variety of cellular tissues and organs (eg, tendons,
ligaments, skin). It is also the major organic component in bones and significantly contributes to overall bone strength. Defects in type I collagen can occur in osteogenesis imperfecta, an autosomal dominant disorder that may present with fragile bones and ligament laxity.
Amiodarone
Amiodarone is a class III antiarrhythmic drug used for the management of a variety of supraventricular and ventricular arrhythmias. Class III drugs predominantly block potassium channels and inhibit the outward potassium currents during phase 3 of the cardiac action potential, prolonging repolarization and total action potential duration (APD).
The QT interval represents the time required for ventricular depolarization and repolarization; it can be regarded as a rough estimate of the APD. Drugs that significantly prolong the cardiac action potential (eg, class IA and III antiarrhythmics) cause prolongation of the QT
interval. Prolonged QT is associated with a form of ventricular tachycardia called torsades de pointes (polymorphic ventricular tachycardia). However, unlike other drugs that cause QT prolongation, amiodarone has very little risk of inducing torsades de pointes. This effect is thought to be due to it having a more homogeneous effect on ventricular repolarization compared to other drugs (ie, less QT dispersion).
Formation of aorta, pulmonary trunk and interventricular septum
The clinical and echocardiographic findings in this cyanotic neonate are consistent
with transposition of the great arteries (TGA). The etiology of TGA is due to linear (rather than spiral) development of the aorticopulmonary septum in utero, resulting in an anteriorly positioned aorta connected to the right ventricle and a posteriorly positioned pulmonary artery connected to the left ventricle. Therefore, pulmonary and systemic circulations are abnormally separated and exist as 2 parallel circulations.
TGA is incompatible with life unless there is another coexisting connection, such as a patent foramen ovale, septal defect, or patent ductus arteriosus (PDA), to allow mixing of oxygenated pulmonary circulation blood with the systemic circulation. Patients may be normal on initial presentation but will become cyanotic, tachypneic, and tachycardic as the PDA (machine-like murmur) begins to close, which typically occurs at age 1-3 days. Elevated lactate in this patient signifies anaerobic metabolism in the presence of poorly oxygenated blood
- Failure of apoptosis of the tissue between the digits can result in syndactyly.
- Failure of fusion is seen in hypospadias (failure of urethral folds to fuse)
- A branchial cleft cyst results from failure of obliteration of the second branchial cleft. The cysts usually present as a draining sinus at the angle of the mandible.
- The membranous portion of the interventricular septum results from proliferation of endocardial cushions. Failure of this proliferation leads to a membranous ventricular septal defect (VSD) that can present as a holosystolic murmur if small, or cyanosis if large. However, the aorta should lie posterior to the pulmonary artery (the normal configuration).
- Failure of conotruncal septation results in persistent truncus arteriosus, which can present with cyanosis and respiratory distress, although an echocardiogram will show a single arterial trunk overriding a large VSD.
Digoxin
Digoxin is a positive inotropic agent that provides symptomatic relief in patients with acute decompensated heart failure due to left ventricular systolic dysfunction. It also increases parasympathetic tone and slows conduction through the atrioventricular node, which can help improve cardiac function in patients with a rapid ventricular rate.
Digoxin directly inhibits the Na-K-ATPase pump in myocardial cells, leading to decreased sodium efflux and an increase in intracellular sodium levels. The decreased transmembrane sodium gradient reduces the forward activity of the sodium-calcium exchanger, leading to a
secondary decrease in calcium efflux from the cells. Increased intracellular calcium concentration stimulates the binding of calcium to troponin C and subsequent actin-myosin cross-bridge formation, resulting in improved myocyte contractility and left ventricular systolic function.
- Cyclic AMP (cAMP), an important second messenger in excitation-contraction coupling, is formed from ATP by adenylyl cyclase. Adenylyl cyclase activity is stimulated by beta agonists and inhibited by acetylcholine. cAMP increases the conductance of the calcium channels in the sarcoplasmic reticulum. As a result, more calcium is available to bind to troponin C, strengthening the force of contraction.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) occurs due to a direct insult to the myocardium that leads to a decrease in contractile function of one or both ventricles with a consequent increase in ventricular cavity size. It can result from a variety of causes, including viral infection, chemical toxicity (eg, doxorubicin, alcohol, cocaine), and infiltrative disease (late manifestation). Patients typically have symptoms of decompensated heart failure (eg, fatigue, dyspnea, orthopnea). In addition, the left ventricular structural changes place these patients at risk for sudden cardiac death due to ventricular arrhythmia.
Patients are classified as having idiopathic DCM when no apparent cause of DCM is identified on routine clinical evaluation. A large percentage of these patients likely have
a familial (inherited) cause. Familial DCM can result from a variety of genetic mutations affecting either sarcomere (ie, the contractile apparatus) or nonsarcomere proteins. Truncating mutations (usually nonsense mutations) affecting the TTN gene, which encodes for the sarcomere protein titin, are the most common cause of familial DCM. Titin is an elastic protein that anchors the beta-myosin heavy chain to the Z-discs and likely contributes to passive myocardial tension; absence of complete titin proteins leads to myocardial dysfunction.
TTN gene mutations follow autosomal dominant inheritance; however, they have incomplete penetrance, leading to delayed or absent clinical manifestations in some family members.
- Arrhythmogenic right ventricular cardiomyopathy is characterized by fibrosis and scarring of right ventricular myocardium, which predisposes to ventricular arrhythmias and sudden cardiac death. The disease results from impaired desmosome function due to mutations in genes encoding desmosomal proteins (eg, plakoglobin, desmoplakin).
- Familial bicuspid aortic valve disease has been associated with a mutation affecting the NOTCH1 gene that encodes transcription regulatory proteins.
- Hypertrophic cardiomyopathy is a common cause of sudden cardiac death in young patients. It is inherited in an autosomal dominant pattern and most commonly involves mutations in the genes encoding beta-myosin heavy chain or myosin-binding protein C.
- Restrictive cardiomyopathy most commonly results from infiltrative disease (eg, amyloidosis, hemochromatosis, sarcoidosis); however, familial disease does occur. Familial restrictive cardiomyopathy typically involves mutations in genes encoding one of several sarcomere or cytoskeletal proteins, but mutations of the TTN gene are not a well-established cause.