Синдром Марфана — это состояние, которое поражает соединительную ткань. Соединительная ткань скрепляет ваше тело и обеспечивает поддержку многих структур по всему телу.
При синдроме Марфана соединительная ткань ненормальна. В результате поражаются несколько систем организма, включая сердце и кровеносные сосуды, кости, сухожилия, хрящи, глаза, кожу и легкие.
Кого поражает синдром Марфана?
Синдром Марфана встречается довольно часто, поражая 1 из 10 000–20 000 человек. Он был обнаружен у людей всех рас и этнических групп.
СИМПТОМЫ И ПРИЧИНЫ
Что вызывает синдром Марфана?
При синдроме Марфана возникает дефект в гене, который кодирует структуру фибриллина и эластических волокон, основного компонента соединительной ткани. Этот ген называется фибриллин-1 или FBN1 .
В большинстве случаев синдром Марфана передается по наследству. Паттерн называется «аутосомно-доминантным», что означает, что он встречается в равной степени у мужчин и женщин и может быть унаследован только от одного родителя с синдромом Марфана. Люди с синдромом Марфана имеют 50-процентный шанс передать заболевание каждому из своих детей.
В 25% случаев дефект нового гена возникает по неизвестной причине. Синдром Марфана также называют генетическим заболеванием с «вариабельной экспрессией», потому что не все люди с синдромом Марфана имеют одинаковые симптомы, и у некоторых людей симптомы могут быть хуже, чем у других.
Синдром Марфана является врожденным заболеванием, однако диагноз обычно не устанавливается до подросткового или юношеского возраста.
Каковы симптомы синдрома Марфана?
Иногда синдром Марфана протекает настолько легко, что сразу заметны лишь немногие симптомы. В большинстве случаев симптомы становятся очевидными, когда с возрастом происходят изменения в соединительной ткани.
Поскольку синдром Марфана влияет на вашу соединительную ткань, он может повлиять на все ваше тело, включая вашу скелетную систему, сердце и кровеносные сосуды, глаза, кожу и органы.
Внешность
Физические особенности включают в себя:
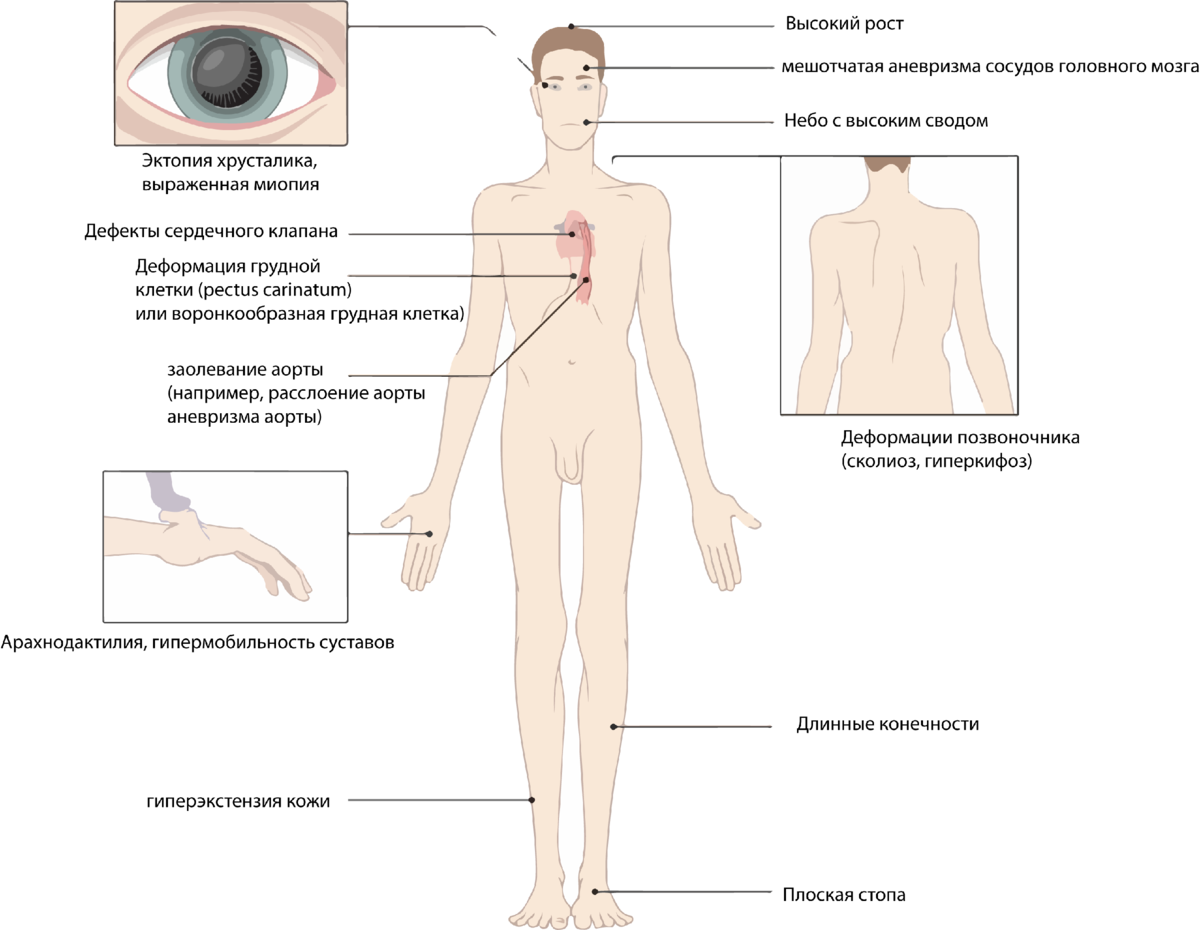
- Длинное, узкое лицо.
- Высокое телосложение и худощавое телосложение.
- Руки, ноги, пальцы рук и ног могут показаться слишком длинными для остального тела.
- Кривой позвоночник. Сколиозом страдают 60% людей с синдромом Марфана.
- Грудина (грудная кость), которая может либо торчать, либо иметь углубления.
- Суставы слабые и легко вывихиваются.
- Плоскостопие.
Стоматологические проблемы
Стоматологические проблемы включают в себя:
- Скученные зубы.
- Узкое, выше, чем в норме, арочное небо (крыша рта).
Проблемы с глазами
Более половины всех людей с синдромом Марфана имеют проблемы со зрением. К ним относятся:
- Близорукость (нечеткость предметов вдали).
- Подвывих хрусталика (хрусталик глаза смещается от своего типичного положения).
- Катаракта.
- Разница в форме глаза.
- Отслойка сетчатки.
- Глаукома.
Проблемы с сердцем и сосудами
Около 90% людей с синдромом Марфана развивают изменения в сердце и кровеносных сосудах. Изменения, которые могут развиться, включают:
- Аневризма аорты. Стенки аорты, главной артерии, несущей кровь от сердца к остальным частям тела, становятся слабыми, выпячиваются и могут разорваться (лопнуть). Чаще всего это происходит в корне аорты (точке, где аортальная артерия выходит из сердца).
- Расслоение аорты. Это разрыв во внутреннем слое трех слоев стенки аорты. Разрыв позволяет крови проникнуть в средний слой, что расширяет разрыв и приводит к дальнейшему разделению и, возможно, разрыву стенки. Это может быть фатальным.
- Проблемы с сердечным клапаном. Синдром Марфана может привести к ослаблению и растяжению ткани клапана. Это приводит к тому, что клапаны не закрываются плотно, вызывая утечки и обратный ток крови. Сердцу часто приходится работать больше, когда клапаны не работают должным образом. Обычно поражается митральный клапан.
- Увеличенное сердце. Сердечная мышца со временем может увеличиваться и ослабевать, вызывая кардиомиопатию, даже если сердечные клапаны не протекают. Состояние может прогрессировать до сердечной недостаточности.
- Аномальный сердечный ритм. У некоторых людей с синдромом Марфана может возникать аритмия. Часто это связано с пролапсом митрального клапана.
- Аневризмы головного мозга. Люди с болезнью Марфана могут иметь в анамнезе внутричерепное кровотечение из-за разрыва аневризмы головного мозга.
Изменения легких
Изменения легочной ткани, возникающие при синдроме Марфана, повышают риск:
- Астма.
- Эмфизема.
- Хроническая обструктивная болезнь легких (ХОБЛ).
- Бронхит.
- Пневмония.
- Спадение легкого (пневмоторакс).
Изменения кожи
Кожа может стать менее эластичной, что приведет к появлению растяжек даже без изменения веса.
ДИАГНОСТИКА И ТЕСТЫ
Как диагностируется синдром Марфана?
Поскольку синдром Марфана может поражать ткани по всему телу, для подтверждения диагноза и разработки плана лечения может быть привлечена группа врачей.
Во-первых, они изучат вашу историю болезни, проведут медицинский осмотр для поиска типичных признаков или результатов, связанных с Марфаном, зададут вопросы о симптомах, которые вы испытываете, и соберут информацию о членах семьи, у которых могли быть проблемы со здоровьем, связанные с синдром.
Тесты для оценки изменений в сердце, кровеносных сосудах и проблем с сердечным ритмом могут включать:
- Рентген грудной клетки, чтобы увидеть границы вашего сердца.
- Электрокардиограмма (ЭКГ) для проверки частоты сердечных сокращений и ритма.
- Эхокардиограмма для выявления проблем с сердечным клапаном, осмотр сердца на предмет расширения или утолщения желудочков, а также осмотр аорты на предмет расширения, расслоения (разрывов) или аневризм.
Если ваш лечащий врач не может увидеть срезы аорты на эхокардиограмме или считает, что расслоение уже произошло, вам может потребоваться дополнительное обследование.
К ним относятся:
- Чреспищеводное эхо (ЧПЭ).
- Магнитно-резонансная томография (МРТ).
- Компьютерная томография (КТ).
Часто также требуется КТ или МРТ для проверки дуральной эктазии. Дуральная эктазия представляет собой выпячивание слизистой оболочки позвоночного столба. Часто это не вызывает никаких симптомов, но у некоторых людей может быть связано с болью в спине. Эктазия твердой мозговой оболочки помогает подтвердить диагноз синдрома Марфана, но также может возникать при других заболеваниях соединительной ткани.
Анализ крови может помочь диагностировать синдром Марфана. Этот генетический тест ищет изменения в FBN1, гене, ответственном за большинство случаев синдрома Марфана.
Консультант-генетик должен пересмотреть ваше генетическое тестирование, потому что результаты теста FBN1 не всегда очевидны. Анализы крови также можно использовать для диагностики других генетических мутаций, таких как синдром Лойса-Дитца, который вызывает физические симптомы, сходные с синдромом Марфана.
ЛЕЧЕНИЕ
Как лечится синдром Марфана?
Если у вас синдром Марфана, вам потребуется план лечения, соответствующий вашим проблемам со здоровьем. Некоторым людям может не понадобиться никакого лечения — достаточно регулярных последующих визитов к лечащему врачу. Другим могут потребоваться лекарства или хирургическое вмешательство. Подход зависит от того, какие части тела поражены, и тяжести вашего состояния.
Лекарства
Лекарства не используются для лечения синдрома Марфана, но их можно использовать для предотвращения или контроля осложнений. Лекарства могут включать:
- Бета-блокаторы: Бета-блокаторы улучшают способность вашего сердца расслабляться и уменьшают силу сердцебиения и давление в артериях. Это предотвращает или замедляет расширение аорты. Терапию бета-блокаторами следует начинать в раннем возрасте. Если вы не можете принимать бета-блокаторы из-за астмы или побочных эффектов, врач может назначить вам блокатор кальциевых каналов.
- Блокаторы рецепторов ангиотензина: Блокаторы рецепторов ангиотензина (БРА) используются для лечения высокого кровяного давления и сердечной недостаточности. Недавние клинические испытания показали, что БРА помогают замедлить расширение аорты так же, как и бета-блокаторы.
Операция
Целью операции при синдроме Марфана является предотвращение расслоения или разрыва аорты и лечение проблем с клапаном.
Решение о хирургическом вмешательстве основывается на:
- Размер вашей аорты.
- Ожидаемый нормальный размер аорты.
- Скорость роста аорты.
- Ваш возраст, рост и пол.
- Семейный анамнез расслоения аорты.
Для замены увеличенного участка аорты трансплантатом можно использовать два хирургических метода:
- Традиционный метод: аорта заменяется трансплантатом, а аортальный клапан заменяется механическим клапаном.
- Модифицированный метод реимплантации с сохранением клапана: аорта заменяется трубчатым трансплантатом, а ваш собственный аортальный клапан возвращается на место. Клапаносохраняющий метод используется, когда это возможно, и должен выполняться опытным хирургом.
Если вам нужна операция, вы должны выбрать больницу, которая имеет опыт в этом типе хирургии. Лучшее понимание синдрома Марфана, более раннее выявление, тщательное последующее наблюдение и более безопасные хирургические методы дают людям лучшие результаты.
Вам может потребоваться лечение проблем, которые синдром Марфана вызывает в других частях тела. Врачи, специализирующиеся на легких, костях и глазах, могут помочь вам с проблемами в этих областях.
ПЕРСПЕКТИВЫ/ПРОГНОЗ
Какова продолжительность моей жизни с синдромом Марфана?
Благодаря достижениям медицины (особенно операциям на сердце) продолжительность жизни людей с синдромом Марфана начала расти в конце 1970-х годов. Раньше ожидаемая продолжительность жизни составляла 32 года. Сегодня некоторые люди с синдромом Марфана могут дожить до 72 лет. Диагноз лучше всего ставить в молодом возрасте, потому что болезнь может прогрессировать и представлять множество рисков.
Что я могу сделать, чтобы оставаться максимально здоровым, если у меня синдром Марфана?
Поскольку синдром Марфана может поражать различные части вашего тела, важно регулярно посещать врачей, которые могут помочь вам с проблемами в областях, затронутых вашим телом.
- Последующее наблюдение: Обычные последующие визиты должны включать осмотры сердца и кровеносных сосудов, глаза и скелета, особенно в период роста. Ваши врачи сообщат вам, как часто вам нужны эти встречи.
- Активность: рекомендации по активности варьируются в зависимости от степени заболевания и симптомов. Большинство людей с синдромом Марфана могут участвовать в той или иной физической и/или развлекательной деятельности. Если у вас расширена аорта, вам, вероятно, следует избегать высокоинтенсивных командных видов спорта, контактных видов спорта и тяжелых упражнений (таких как тяжелая атлетика). Спросите своего врача о рекомендациях по активности для вас.
- Беременность. Перед беременностью следует пройти консультацию у генетика, поскольку синдром Марфана является наследственным заболеванием. Беременные люди с синдромом Марфана относятся к группе высокого риска. Если ваша аорта нормального размера, риск расслоения ниже, но все же существует. Те, у кого есть даже небольшое увеличение, подвергаются более высокому риску, а стресс во время беременности может вызвать расширение быстрее. Во время беременности вам потребуется тщательное наблюдение с частыми проверками артериального давления и ежемесячными эхокардиограммами. Если есть быстрое увеличение или аортальная регургитация, может потребоваться постельный режим или хирургическое вмешательство. Ваш врач обсудит с вами наилучший метод родоразрешения.
- Профилактика эндокардита: Если у вас была операция на клапане, у вас повышен риск бактериального эндокардита. Это инфекция сердечных клапанов или ткани, которая возникает, когда бактерии попадают в кровоток. Чтобы снизить риск эндокардита, перед стоматологическими или хирургическими процедурами следует назначать антибиотики. Проконсультируйтесь с врачом о типе и количестве антибиотиков, которые вам следует принимать.
Заключение
Узнав, что у вас есть генетическое заболевание, такое как синдром Марфана, вы можете быть озабочены изменением своего образа жизни, оплатой лечения, необходимостью хирургического вмешательства и постоянным медицинским наблюдением. Для того, чтобы получить точную информацию и оценку рисков, важно обратиться за медицинской помощью к специалисту, который имеет опыт лечения данного синдрома. Кроме того, вы можете беспокоиться о возможном риске передачи заболевания вашим будущим детям, поэтому рекомендуется обратиться к врачу-генетику, чтобы получить необходимую информацию и поддержку.