
Словарь:
АННОТАЦИЯ ГЕНОМА- маркировка генов и других объектов внутри них.
ГЕНОМ - наследственный материал, заключенный в клетке организма и необходимый для построения и поддержания его жизнедеятельности. Раздел молекулярной генетики, занимающийся геномом и генами организмов, принято называть геномикой.
ДНК - дезоксирибонуклеиновая кислота, макромолекула, обеспечивающая хранение, передачу из поколения в поколение и реализацию генетической программы у многих организмов.
ДНК-БИБЛИОТЕКА (ГЕНОМНАЯ БИБЛИОТЕКА)- набор ДНК-фрагментов всего генома организма. Эти ДНК-фрагменты фланкированы идентичными ДНК-адаптерами и содержат различные вставки генома.
ДНК-АДАПТЕРЫ - небольшие фрагменты ДНК известной последовательности, фланкирующие ДНК-библиотеку. Используются как участки, с которых начинается амплификация или секвенирование ДНК-библиотеки.
МЕТИЛИРОВАНИЕ ДНК - своеобразная модификация молекулы ДНК путем присоединении метильной группы (-CH3) к цитозину в составе CpG-динуклеотида. Один из способов регуляции работы генома без изменения нуклеотидной последовательности ДНК.
НУКЛЕОТИДЫ (НУКЛЕОЗИДФОСФАТЫ)- низкомолекулярные вещества (мономеры), составляющие сложные биологические полимеры: соответственно РНК или ДНК. Нуклеотиды, составляющие ДНК: аденин (A), тимин (T), гуанин (G) и цитозин (C). В РНК вместо тимина присутствует урацил (U).
ПЦР (ПОЛИМЕРАЗНАЯ ЦЕПНАЯ РЕАКЦИЯ)- молекулярно-биологический метод, позволяющий добиться колоссального (до 1012 раз) увеличения или амплификации числа копий определенного фрагмента ДНК in vitro.
РНК - рибонуклеиновая кислота, макромолекула, образующаяся у многих организмов в результате считывания с ДНК (транскрипции), необходима в процессе синтеза белков, а также многочисленных регуляторных процессов в клетке. Также ответственна за хранение, передачу из поколения в поколение и реализацию генетической программы у некоторых вирусов.
РЕСЕКВЕНИРОВАНИЕ - повторное определение последовательности ДНК организма с уже расшифрованным геномом (применяется, например, при поиске вариантов, связанных с наследственными заболеваниями у человека).
СЕКВЕНИРОВАНИЕ ДНК ИЛИ РНК - определение первичной последовательности нуклеотидов в составе макромолекул, несущих наследственную информацию.
СЕКВЕНИРОВАНИЕ DE NOVO - секвенирование нового для науки (ранее не прочитанного) генома.
ЧТЕНИЯ (РИДЫ, READS) - фрагменты ДНК (длиной от 25 до 20 000 нуклеотидов), считываемые с генома при помощи специальной машины — секвенатора.
ЭМУЛЬСИОННАЯ ПЦР (ЭПЦР) - ПЦР, проводимая в эмульсии, где в каждой капельке масла амплифицируется единичная молекула ДНК (фрагмент ДНК).
ЭПИГЕНОМИКА - один из разделов геномики, посвященный изучению изменения экспрессии генов, вызванного механизмами, не затрагивающими последовательности ДНК, например, через метилирование ДНК.
Почти четверть века назад в Соединенных Штатах стартовал грандиозный по своему масштабу научный проект, посвященный определению последовательности генома человека. В течение тринадцати лет многочисленные исследовательские группы по всему миру работали над определением полной последовательности генома человека. Почти три миллиарда долларов, потраченные на этот проект, открыли перед исследователями замечательные перспективы. Используя полученные данные, появилась возможность искать и находить участки ДНК, связанные с генетически обусловленными заболеваниями. Такие заболевания могут быть моногенными (вызываемых отказом единственного гена) или многофакторными, и вызвать их может широкий спектр изменений генома, многие из которых до сих пор неизвестны (напр., Альцгеймера и Паркинсона).
Геномные исследования позволяют решать массу задач как прикладного, так и фундаментального плана. Благодаря им разрабатываются новые лекарства и продукты, они же позволяют проникнуть в глубокую историю человечества.
Методы секвенирования первого поколения
Самый популярный и надежный из них — секвенирование по Сэнгеру — позволяет «считывать» последовательности до 1000 пар оснований (п.о.) и используется для небольших фрагментов генома/генов или для валидации результатов более современного секвенирования нового поколения (next-generation sequencing, NGS), где размер одного прочитанного фрагмента варьирует от 25 до 500 п.о. В отличие от секвенирования по Сэнгеру, методы NGS используют для глубокого (многократного) прочтения генетического материала, которое необходимо, например, для ресеквенирования и сборки новых геномов (de novo), транскриптомных и эпигеномных исследований. Они значительно производительнее, позволяя одновременно считывать миллионы и даже миллиарды коротких фрагментов. Такой рост производительности привел к возможности определения последовательности сразу десятков геномов (в зависимости от их размера) за один запуск прибора.
Основные, как я писала уже:
- Ion Proton и Ion Personal Genome Machine (Thermo Fisher Scientific) — технология ионного полупроводникового секвенирования;
- MiSeq и NovaSeq (Illumina) — технология секвенирования на молекулярных кластерах с использованием флуоресцентно меченых нуклеотидов;
- MinION, GridION X5, PromethION и SmidgION (Oxford Nanopore Technologies) — нанопоровое секвенирование.
Все современные секвенирующие платформы отличаются от метода секвенирования по Сэнгеру тем, что не требуют этапа клонирования фрагментов ДНК. Это экономит рабочее время и позволяет избежать ряда проблем с клонированием АТ-богатых участков. Общий принцип пробоподготовки для большинства современных (NGS) секвенаторов включает фрагментирование ДНК, привязку к субстрату, амплификацию фрагментов с помощью ПЦР (в одномолекулярном секвенировании от ПЦР удалось отказаться) и последующее считывании последовательности НК. В отличие от метода секвенирования по Сэнгеру, современные платформы обеспечивают параллельное проведение миллиардов реакций в малых объемах, что позволяет получить намного больший объем информации на выходе.
Собственно, Сэнгер предложил два способа - 1. «плюс-минус» метод секвенирования ДНК - Методику секвенирования ДНК с использованием радиоактивно меченых нуклеотидов и ДНК-полимеразы (или фрагмента Кленова ДНК-полимеразы I): ПЦР с меткой одного дезоксинуклеотида по α-положению фосфата (32P). В «плюс»-системе проводят четыре ПЦР-реакции в присутствии каждого из четырех типов дезоксинуклеозидтрифосфатов; параллельно в «минус»-системе проводят четыре ПЦР-реакции в отсутствии каждого из них. Далее результаты визуализируют с помощью электрофореза.
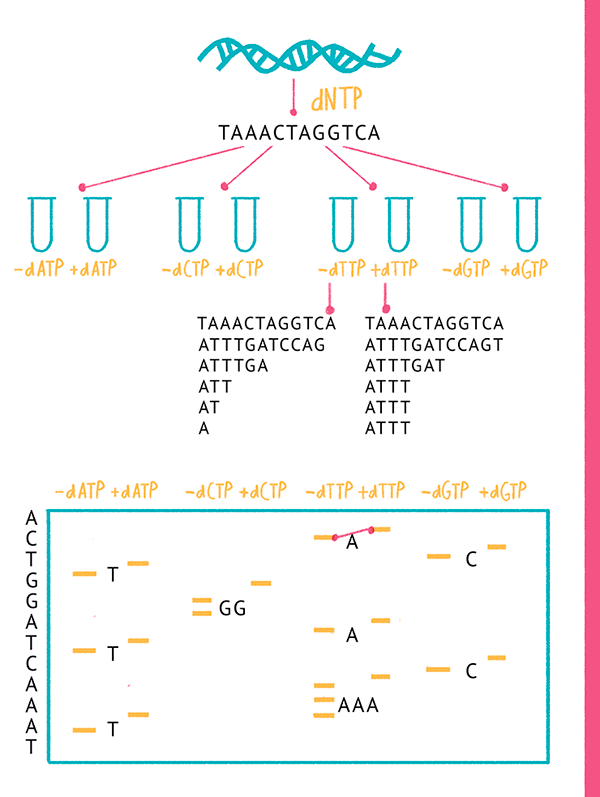
2. метод «терминаторов» или «обрыва цепи» - в реакционную смесь добавляют аналоги привычных нуклеотидов (дидезоксинуклеозидтрифосфаты), включение которых в синтезируемую цепь приводит к невозможности ее дальнейшего синтеза (терминации), а по образовавшемуся «обломку» можно установить последнюю букву секвенируемого фрагмента ДНК.
Затем радиоактивные метки заменили флуоресцентными.
По Сэнгеру работают капиллярные секвенаторы. Производительность — 4, 8, 24, 48 и 96 образцов. Они предназначены для секвенирования относительно небольших фрагментов ДНК длиной до 1000 нуклеотидов, характеризуются высокой точностью и используются для генотипирования человека, животных и других организмов (в том числе в медицине — трансплантологии, перинатологии, онкологии, фармакогенетике и др.), для выявления точечных мутаций, делеций и инсерций, для валидации мутаций, выявленных NGS, для анализа метилирования ДНК, для идентификации личности в криминалистике и др.
Как отмечалось, из первого поколения есть ещё метод химической деградации, разработанный Максамом и Гилбертом. Но он не получил дальнейшего распространения из-за быстрого развития энзимологии.
Итак, методы первого поколения используют для:
- секвенирования отдельных участков генома с целью анализа мутаций и полиморфизмов;
- идентификации вирусов и организмов (бактерий, растений, грибов и животных);
- валидации данных, полученных на платформах секвенирования нового поколения (NGS);
- микросателлитного анализа;
- анализа делеций и инсерций (малых и протяженных).
Пиросеквенирование - «секвенирование путем синтеза». Технологию предложил в 1996 году Пол Нирен с коллегами из Королевского технологического института в Стокгольме. И она дала начало новым подходам в секвенировании. Ее коммерциализировали (2005 год) и воплотили в приборе GS FLX, 454 производства Roche (2008 год). Этим методом можно определять нуклеотидную последовательность фрагментов геномной ДНК размером 300–500 пар оснований (п.о.). Основной смысл этого типа секвенирования заключается в последовательном синтезе ДНК на ДНК-фрагментах изучаемого организма в специальных пиколитровых «реакторах».
В ходе синтеза дочерней цепочки ДНК детектируют пирофосфаты, высвобождающиеся при включении нуклеотида в синтезируемую на матрице (участке молекулы ДНК, служащим матрицей для синтеза) комплементарную цепь. К обоим концам фрагментированной ДНК «пришивают» ДНК-адаптеры (данная конструкция называется ДНК-библиотекой), необходимые для эмульсионной ПЦР (эПЦР) на магнитных сферах и последующего секвенирования.
Готовые ДНК-библиотеки иммобилизуют на магнитных сферах. Затем магнитные сферы с нанесенной на них клональной библиотекой доставляют на проточную ячейку, где в присутствии праймера, дезоксинуклеотидтрифосфатов и ферментов — ДНК-полимеразы, люциферазы, АТФ-сульфурилазы — происходит циклический синтез новой цепи.
Во время цикла пиросеквенирования при образовании фосфодиэфирной связи между матричной цепочкой ДНК и нуклеотидом синтезируемой цепи выделяется пирофосфат, который запускает каскад химических реакций, приводящих к выделению АТФ, необходимой для реакции окисления люциферина с выделением кванта света, который фиксируют аналоговой интегральной микросхемой (ПЗС-матрицей), состоящей из светочувствительных фотодиодов. Нуклеотиды, не вовлеченные в синтез новой цепи, удаляют из проточной ячейки, и начинается следующий реакционный цикл, в ходе которого добавляют дезоксинуклеотидтрифосфат другого типа.
Технология секвенирования на молекулярных кластерах с использованием флуоресцентно меченых нуклеотидов
Этот метод начали разрабатывать еще в середине 90-х годов прошлого века химики Шанкар Баласубраманиан и Дэвид Кленерман из Кембриджа, изучавшие работу ДНК-полимеразы на молекулярном уровне, используя флуоресцентно меченые нуклеотиды и ДНК-матрицу, иммобилизованную на поверхности. Подход был приобретен компанией Illumina. Сейчас платформа продолжает развиваться, и потребителям предлагают новые линейки приборов.
К обоим концам предварительно фрагментированной ДНК лигируют адаптеры, необходимые для ПЦР и последующего секвенирования на молекулярных кластерах. Полученные ДНК-библиотеки иммобилизуют на поверхности проточной ячейки, где и проводят циклический процесс секвенирования. Реакционная смесь для синтеза комплементарной ДНК подается на поверхность проточной ячейки и содержит ферменты, олигонуклеотиды, а также четыре типа флуоресцентно меченых дезоксинуклеозидтрифосфатов. После включения в синтезируемую цепь ДНК нуклеотида-терминатора идентифицируют с помощью ПЗС-матрицы как тип включенного нуклеотида, так и его положение. Затем терминирующая группа и флуоресцентная краска отщепляются от нуклеотида, и цикл синтеза повторяется. Эта серия шагов продолжается определенное количество раз, число которых задает пользователь.
Размер чтений, получаемых с секвенатора, может достигать 300 п.о. (прибор Illumina MiSeq). Кроме того, серия секвенаторов 2017 года NovaSeq позволяет определять последовательность до 48 геномов человека за один запуск прибора.
Секвенирование «нового поколения» — next-generation sequencing (NGS)
началось с внедрения последних двух описанных методов, но затем стали появляться новые предложения.
Секвенирование «нового поколения» применяется как для анализа геномов организмов, для которых уже доступен референсный геном (ресеквенирование), так и для того, чтобы впервые расшифровать геном организма (секвенирование de novo).
Для ресеквенирования успешно используют платформы, генерирующие большое количество коротких чтений (секвенируемых фрагментов ДНК), поскольку даже относительно короткие фрагменты ДНК успешно картируются (картирование, или выравнивание, — процесс биоинформатического поиска расположения конкретного короткого фрагмента в полной геномной последовательности) на референсный геном (последовательность ДНК в цифровом виде, составленную учеными как общий репрезентативный пример последовательности генома конкретного вида) при биоинформатическом анализе данных. Такие выравненные чтения могут использоваться для поиска однонуклеотидных полиморфизмов (SNPs), малых делеций и инсерций или других структурных изменений в геноме.
Что касается секвенирования de novo ранее не прочитанных геномов, то использование коротких чтений сильно усложняет сборку, особенно в случае больших по размеру и сложно устроенных геномов эукариот (например, полиплоидных геномов). В этих случаях используют комбинированный подход — сочетание платформ, генерирующих как короткие, так и длинные чтения.
Технология циклического лигазного секвенирования была разработана группой Джорджа Макдональда Черча. Она использует метод лигирования (формирование химических связей между нуклеотидами при помощи специального фермента — лигазы). Данный подход к секвенированию НК коммерциализировали в 2006 году, и приборы, известные под брендом SOLiD, уже длительное время представлены на рынке (первоначально развитием этой системы секвенирования занималась компания Applied Biosystems, а затем Life Technologies и Thermo Fisher Scientific).
Суть метода заключается в определении нуклеотидной последовательности фрагментов геномной ДНК размером 25–75 п.о. К обоим концам предварительно фрагментированной ДНК лигируют адаптеры, необходимые для эПЦР на магнитных сферах и последующего секвенирования на проточной ячейке.
Магнитные сферы с нанесенной на них клональной библиотекой помещают на проточную ячейку, где и происходит секвенирование с помощью лигирования восьминуклеотидных зондов, несущих четыре различных флуорофора на 5’-конце. Флуоресценция считывается с помощью специальной камеры после каждого цикла секвенирования и, затем переводится в последовательность нуклеотидов.
Ионное полупроводниковое секвенирование
Ионное полупроводниковое секвенирование, основанное на технологии PostLightTM, разработано компанией Ion Torrent и в настоящее время применяется в приборах, реализуемых Thermo Fisher Scientific — Ion S5 / Ion S5 XL, Ion Proton, Ion Personal Genome Machine (PGM).
Технология, предлагаемая в этой приборной линейке, основана на использовании полупроводниковых микрочипов для секвенирования. Суть этого подхода весьма проста и заключается в регистрации локального изменения рН на микрочипе в момент удлинения синтезируемой цепи ДНК-полимеразой на ДНК-матрице.
Пробоподготовка (приготовление ДНК-библиотек) напоминает таковую при циклическом лигазном секвенировании. Первоначально ДНК фрагментируют, затем к концам полученных фрагментов лигируют специфические ДНК-адаптеры, необходимые для эмульсионной ПЦР на магнитных сферах и последующего секвенирования.Пробоподготовка (приготовление ДНК-библиотек) напоминает таковую при циклическом лигазном секвенировании. Первоначально ДНК фрагментируют, затем к концам полученных фрагментов лигируют специфические ДНК-адаптеры, необходимые для эмульсионной ПЦР на магнитных сферах и последующего секвенирования.
Секвенирование третьего поколения
Одномолекулярное секвенирование
SMRT-секвенирование (single molecule real time sequencing), предложенное сотрудниками компании Pacific Biosciences, не только позволило отказаться от проведения полимеразной цепной реакции при пробоподготовке, но и дало возможность наблюдать за работой ДНК-полимеразы, наращивающей синтезируемую цепь, в реальном времени.
Создание платформы не только решило проблему ПЦР-дупликатов, но и значительно увеличило длину чтений, что крайне важно при сборке геномов de novo. Суть метода заключается в определении нуклеотидной последовательности фрагментов геномной ДНК размером до 20 000 п.о. с лигированными к их концам специфическими ДНК-адаптерами, необходимыми для последующего секвенирования.
Сама реакция секвенирования молекул ДНК проходит в специальных ячейках (SMRT-ячейки) на прозрачной (кремниевой) подложке, с напыленным на нее слоем алюминия. В основе метода лежит использование технологии Zero-mode waveguide (ZMW).
Сквозь дно в ячейку подается свет, однако благодаря особенностям ее строения, пучок фотонов не рассеивается, а освещает только конкретную часть (на подложке), где закреплена молекула phi29 ДНК-полимеразы. Эта полимераза была выбрана в качестве «считывающего» фермента благодаря своей высокой точности, скорости синтезирования дочерней цепи и эффективной работе с нуклеотидами, несущими флуоресцентную метку.
Смысл SMRT-секвенирования схож с описанными ранее методами NGS — ДНК-полимераза достраивает вторую цепь исследуемой молекулы ДНК, используя нуклеотиды, меченные различными флуоресцентными метками, которые регистрируют при помощи конфокальной микроскопии.
Разработка другого способа одномолекулярного секвенирования (коммерциализированного к настоящему времени) началась в конце прошлого века, когда группа американских ученых наглядно продемонстрировала возможность побуждать молекулы ДНК и РНК проходить сквозь ионный канал диаметром 2,6 нм в двуслойной липидной мембране под воздействием электрического поля. Более того, уже тогда исследователи сумели различать ДНК и РНК, а также оценивать длину входящих в нанопору олигонуклеотидов. Данную технологию коммерциализировала и представила на рынок компания Oxford Nanopore Technologies.
Реакционная камера, в которой проходит процесс считывания последовательности НК, разделена двухслойной мембраной с единичной порой. К камере прикладывается напряжение, вызывающее движение ионов и молекул ДНК или РНК через пору. При прохождении молекулы НК сечение поры (доступное для миграции ионов) уменьшается, в результате чего сила тока падает. Таким образом, считывая изменение силы тока, можно определять тип нуклеотида, проходящего через пору в конкретный отрезок времени.
Использование секвенирования нового поколения позволяет проводить такие проекты как:
- Полногеномный анализ (в том числе, ресеквенирование и секвенирование de novo). Ресеквенирование полных геномов человека в интересах персонализированной медицины или секвенирование ранее не изученных геномов вирусов, бактерий, архей, растений, грибов и животных как с чисто фундаментальными, так и прикладными целями.
- Секвенирование РНК (RNA-Seq), позволяющее оценивать экспрессию генов не только качественно, но и количественно. Существует возможность отдельно оценивать экспрессию кодирующих и регуляторных РНК. Данные методики направлены на изучение работы генома (активности его генов, в том числе генов-регуляторов) в разных клетках, тканях и органах.
- Метагеномное секвенирование — оценка разнообразия микроорганизмов в различных образцах. Позволяет оценивать бактериальное разнообразие в различных средах, например, в кишечнике человека, донных отложениях озера Байкал или в горячих источниках Камчатки.
- Анализ ДНК-белковых взаимодействий (ChIP-Seq) — изучение влияния транскрипционных факторов и других ДНК-связывающих белков на экспрессию генов, а через нее на фенотипические и физиологические особенности клеток, органов и тканей.
- Бисульфитное секвенирование и его модификации (например, RRBS) — оценка метилирования в геноме или его участках. Влияние метилирования регуляторных участков генома на уровень экспрессии генов через подавление их транскрипционной активности.
- Таргетное секвенирование (экзомное секвенирование, секвенирование митохондриальных генов, секвенирование ампликонов). Секвенирование отдельных (выбранных исследователем) участков генома, например, только генов митохондриальной ДНК, кодирующих белки генов или генов, для которых уже описано участие в процессах онкогенеза. Таргетное секвенирование позволяет значительно снизить стоимость эксперимента (из расчета на один образец) и многократно увеличить количество анализируемых образцов.
Медицина
Кстати, о данных секвенирования - один запуск даёт около терабайта данных, то есть необходимы довольно большие хранилища.
Референсная последовательность должна быть получена из:
● RefSeq базы данных Национального центра биотехнологической информации (http://www.ncbi.nlm.nih.gov/RefSeq/) с указанием номера версии или
● из базы данных Locus Reference Genomic (http://www.lrgsequence.org).
Используют базы:
1000 Genomes Project http://browser.1000genomes.org/index. html
dbSNP http://www.ncbi.nlm.nih.gov/snp
Human Gene Mutation Database http://www.hgmd.cf.ac.uk/ac/index. php
MitoMap http://www.mitomap.org/MITOMAP/
предсказательные программы
Структура/функция белка SNPs&GO http://snpsandgo.biocomp.unibo.it/snpsandg o/
Анализ данных
можно почитать тут и посмотреть тут:
Секвенирование всего генома (WGS), также известное как полное секвенирование генома, полное секвенирование генома или секвенирование всего генома, представляет собой процесс определения всей или почти всей последовательности ДНК генома организма за один раз. На него возлагает надежды персонализированная медицина. Есть методы, которые секвенируют определенные подмножества генома – такие методы включают секвенирование всего экзома (1-2% генома) или генотипирование SNP (<0,1% генома).
Проект "Геном человека" стартовал в 1990 году, а расшифровка данных была закончена только в 2022-м. Впрочем, и она была неполной.
В начале 2000-х годов ученым удалось определить последовательность большинства участков — примерно 92% всего генома. Но с расшифровкой оставшейся части пришлось повременить.
Сложность была в том, что эти 8% состояли из большого числа коротких повторяющихся участков. У вас есть сотни участков, неотличимых друг от друга. Как сложить их в структурные единицы? Технологии того времени этого не позволяли. Останавливало ученых и то, что эта ДНК считалась вспомогательной и потому не особенно важной.
Закончить работу удалось лишь спустя два десятка лет, с помощью новейших методов секвенирования, которые использовал научный консорциум Telomere-to-Telomere: Oxford Nanopore DNA и PacBio HiFi. Первый за одно прочтение может покрыть до миллиона "букв" с умеренной степенью точности, второй — около 20 тыс., зато практически без ошибок. Сочетание этих двух методов позволило эффективно и быстро секвенировать оставшиеся части.
Так удалось открыть 99 новых генов, которые кодируют белки, и примерно две тысячи генов-кандидатов, роль которых только предстоит изучить. Часть новооткрытых генов отвечают за иммунную реакцию на вирусы и другие патогены; другие участвуют в том, как клетка реагирует на некоторые лекарства; еще часть отвечает за размер нашего мозга.
Значительная часть этих новых открытий связана с двумя очень важными структурами хромосом: теломерами и центромерами.
Первые — это своего рода колпачки, защищающие ДНК от повреждений при делении клетки. Чем больше делений переживает клетка, тем короче становятся теломеры. Это снижает эффективность их защитной функции. Теломеры связаны со старением организма, поэтому их изучение необходимо для понимания этого процесса.
Что касается центромер, то это центральная структура, с помощью которой соединяются две части (плеча) хромосомы. Центромеры играют важную роль в делении клеток, обеспечивая равномерное распределение генетического материала и не допуская появление клеток с большим или меньшим количеством хромосом, чем нужно.
Как объяснил в своем заявлении один из членов группы, Эван Эйхлер, "полная последовательность генома показывает, что некоторые гены, связанные с мозгом, сильно различаются". Например, у одного человека может быть десять копий определенного гена, а у других — только одна или две. Это может дать много подсказок об индивидуальных различиях в мышлении и психике.
Ученые ожидали, что информация о геноме откроет путь к созданию новых лекарств, обеспечит прорыв в здравоохранении. Однако со временем этот взгляд изменился. Оказалось, что знать роль только человеческих генов недостаточно. Например, здоровье человека определяется и геномом организмов, которые с ним сосуществуют, — бактерий, вирусов, грибков.
Кроме того, важно составлять банки геномов целых человеческих популяций — это дает представление о генетическом разнообразии людей. Например, о том, как влияют на здоровье те или иные вариации (полиморфизмы) одного гена. Этим занимается проект "Тысяча геномов", который нацелен на сбор и расшифровку геномов людей из разных частей планеты. Создаются национальные банки ДНК: исландская компания deCODE genetics, например, собрала генетическую информацию о двух третьих населения Исландии. Есть банк Великобритании.
Эти данные используются для развития персонализированной медицины — индивидуального подхода к терапии на основании генетических данных пациента. Например, на основе генома можно понять, как подействует на раковую опухоль та или иная комбинация препаратов. Анализируя мутации, обнаруженные в геномных данных младенца, можно будет предсказать и предупредить развитие болезни.