Продолжаем наш рассказ о новых молекулярно-биологических методах исследования, которые появились в распоряжении ученых буквально в последние несколько лет. Несмотря на сырость методик и предварительность результатов, уже сейчас с их помощью получены важнейшие данные, впервые раскрывающие механизм шизофрении на молекулярном уровне.
Первую часть текст можно (и даже нужно) прочитать здесь.

Топологическое решение
Все С-методы основаны на принципе лигирования по сближенности (proximity ligation). Если разрезать ДНК в ядрах клеток на очень мелкие фрагменты специальными ферментами рестриктазами (такие фрагменты носят название рестриктных и имеют размер порядка 1 килобазы), а затем сшить (слигировать) их между собой обратно, мы получим сложную смесь новых «химерных» молекул ДНК. Химерными они являются по той причине, что рестриктные фрагменты не сшиваются обратно в первоначальные молекулы хромосомной ДНК, а образуют в каждом отдельном ядре уникальные длинные линейные молекулы.
Эти химерные продукты, как из лоскутков, состоят из перекомбинированных (по сравнению с молекулами изначальной геномной ДНК) рестриктных фрагментов. Порядок, в котором рестриктные фрагменты сшиваются между собой, не совсем случаен. У каждого из них есть определенные предпочтительные партнеры по лигированию. Принцип лигирования по сближенности как раз и заключается в том, что основным фактором, определяющим вероятность сшивания двух рестриктных фрагментов, является их близость между собой в трехмерном пространстве ядра. Далекие друг от друга в линейной последовательности генома фрагменты могут предпочтительно сшиваться между собой, если они сближены, образуя что-то наподобие петли.
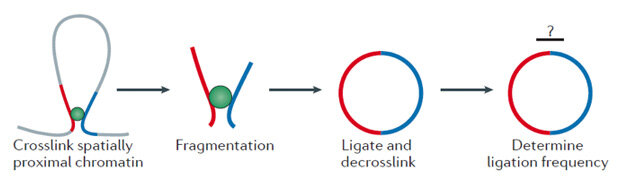
Впервые этот незамысловатый подход был применен в середине 90-х годов для анализа пространственной организации области генома крысы, содержащей ген пролактина. Однако широкое распространение получила модификация, появившаяся в начале 2000-х, в которой для оценки частоты лигирования между двумя определенными рестриктными фрагментами стала использоваться количественная ПЦР. Этот модифицированный метод получил название 3С (chromosome conformation capture). Дальнейшее развитие способов количественной оценки частот лигирования привел к появлению целого семейства С-методов: 4С (circular 3C), 5С (3C carbon copy), Hi-C (high-throughput 3C), ChIA-PET (Chromatin interaction analysis by paired-end tag sequencing) и другие.
С момента появления 3С в науке окончательно утвердилась точка зрения, согласно которой удаленные регуляторные элементы сближены в пространстве (образуя петли) с промоторами регулируемых ими генов. Такие контакты обычно тканеспецифичны и реализуются в клетках, в которых данный ген транскрибируется, или в их непосредственных предшественниках.
Наиболее масштабным С-методом, позволяющим количественно оценивать частоту всех возможных парных комбинаций сшивающихся рестриктных фрагментов генома, является Hi-C. Для подсчета того, с какой частотой каждая из миллионов потенциально возможных комбинаций реализуется (геном разрезается на порядка 106фрагментов, из них можно составить порядка 1012 парных комбинаций), используется высокопроизводительное секвенирование.
Для представления результатов Hi-C часто строятся двумерные графики, называемые тепловыми картами (heat maps) Hi-C, или просто Hi-C картами. На таких графиках вдоль каждой из осей отложены геномные координаты. Соответственно, каждая точка, на плоскости имеющая две координаты, соответствует комбинации двух рестриктных фрагментов. Частота лигирования между двумя этими фрагментами обозначена цветом.
Для Hi-C карт используются разные цветовые шкалы, но наиболее часто встречается красная, в которой белому цвету соответствует отсутствие лигирования между данными рестриктными фрагментами, а красному — максимальная частота лигирования. Таким образом увеличение насыщенности красного цвета отражает увеличение частоты лигирования.
Так как частота лигирования фрагмента a с фрагментом b (точка с координатами a;b на Hi-C карте) тождественно равна частоте лигирования фрагмента b с фрагментом a (точка с координатами b;a на Hi-C карте), в целом карта имеет ось зеркальной симметрии, направленную под углом 45 градусов. Вдоль диагонали обычно располагается область наибольшей частоты контактов, что логично, так как именно здесь находятся пары фрагментов, располагающиеся недалеко друг от друга в линейной последовательности генома. Эти фрагменты всегда имеют тенденцию быть недалеко друг от друга и в пространстве, а следовательно, довольно часто лигироваться между собой.
Иногда на довольно большом расстоянии от диагонали можно увидеть яркие пятна — это и есть пространственно сближенные между собой регуляторные элементы (часто это именно промоторы генов, взаимодействующие со своими УРЭ), чьи координаты можно узнать, опустив перпендикуляры на оси. Далеко не всегда контакты можно различить глазом на Hi-C карте, и тогда для поиска взаимодействий используются дополнительные биоинформатические алгоритмы.
В последние несколько лет с помощью Hi-C и родственных ему методов (несмотря на высокую «эзотеричность») удалось установить много поразительных закономерностей пространственной организации ДНК в клетках млекопитающих. Постепенно мы приходим к пониманию того, какие правила определяют эту организацию и как она связана с регуляцией экспрессии.
Одним из важнейших прикладных применений Hi-C и других C-методов является поиск УРЭ, влияющих на экспрессию тех или иных генов. В последние годы C-методы набирают все большую популярность как инструмент для интерпретации результатов GWAS.
Именно в качестве такого инструмента Hi-C был использован группой ученых из Университета Калифорнии Лос-Анджелес (UCLA). Результаты их работы были описаны в статье, озаглавленной «Chromosome conformation elucidates regulatory relationships in developing human brain», вышедшей в октябре прошлого года в журнале Nature.
С помощью Hi-C учеными была создана карта пространственной организации ДНК в нейронах развивающейся коры головного мозга. Для исследования были взяты клетки коры на 17-18 неделе эмбрионального развития, как раз во время пика активности нейрогенеза. Ткани эмбриональной коры были разделены на кортикальную пластинку (КП), содержащую постмитотические (неделящиеся дифференцированные) нейроны, и герминальную зону (ГЗ), в которой преобладают делящиеся предшественники нейронов.
Пространственная организация ДНК была проанализирована в двух этих клеточных типах по отдельности. Биоинформатический анализ позволил аннотировать тысячи пар контактирующих участков ДНК — как общие для этих клеточных типов, так и уникальные для каждого из них. Одной из главных целей авторов работы, как уже говорилось, было использовать данные о пространственной организации ДНК для интерпретации последнего GWAS на шизофрению, опубликованного в 2014 году.
Среди исследователей довольно давно существует мнение, что по крайней мере некоторые из процессов, нарушаемых у больных шизофренией, проходят как раз в ходе эмбрионального развития мозга. Поэтому авторы статьи посчитали, что их клеточная модель очень хорошо подходит для сопоставления результатов GWAS и данных о пространственной укладке ДНК.
Авторы выбрали около 10 000 SNP, находящихся внутри 108 регионов GWAS и потенциально являющихся каузальными вариантами. Далее они проанализировали паттерн их пространственных контактов. Было обнаружено около 900 генов, взаимодействующих с тем или иным потенциальным каузальным вариантом хотя бы в одном из исследованных клеточных типов. Очевидно, что далеко не все из обнаруженных пар «потенциальный каузальный SNP — взаимодействующий с ним в эмбриональной коре ген» играют какую-либо роль в развитии болезни.
Однако в подтверждение того, что в их данных много информации, ценной для понимания молекулярных основ шизофрении, авторы приводят ряд аргументов.
Во-первых среди обнаруженных генов есть несколько, чья роль в этиологии шизофрении не вызывает сомнений: это уже упоминавшиеся гены DRD2, GRIA1, GRIN2A и CACNA1C. Данные о пространственной укладке показывают, какие именно полиморфизмы внутри регионов GWAS могут влиять на экспрессию этих генов. Кроме того, пул из 900 генов, взаимодействующих с полиморфизмами внутри регионов GWAS, обогащен генами ацетилхолиновых рецепторов (CHRM2, CHRM4, CHRNA2, CHRNA3, CHRNA5 и CHRNB4) и генами, продукты которых принимают участие в нейрогенезе и дифференцировке клеток разных слоев коры мозга (FOXG1, EMX1, TBR1, SATB2, CUX2 и FOXP1).
Очевидно, что такое обогащение не случайно и регуляция по крайней мере некоторых из этих генов нарушена при шизофрении. Важно, что ацетилхолиновые рецепторы уже являются мишенями для разрабатываемых антипсихотических препаратов.
Данные, полученные в этой работе, вероятно, привлекут к ним еще больший интерес.
И наконец, последнее подтверждение значимости своих результатов исследователи нашли, сравнивая пул из обнаруженных ими 900 генов с теми генами, чья экспрессия нарушена в мозге больных шизофренией по данным консорциума CommonMind. Оказалось, что две этих группы генов статистически достоверно пересекаются.
В качестве доказательства того, что их данные являются надежной основой для дальнейших исследований, авторы подтвердили функциональное значение одного из обнаруженных ими пространственных взаимодействий. Для изучения был выбран SNP rs1191551, располагающийся в одном из регионов GWAS и взаимодействующий с геном FOXG1, находящимся на расстоянии 750 килобаз (!!!) от rs1191551.
В предшествующих работах почти не обсуждалась связь этого гена с шизофренией, однако было известно, что он участвует в развитии мозга, а некоторые мутации в нем приводят к тяжелым когнитивным нарушениям. С помощью так называемого люциферазного теста авторы показали, что последовательность, содержащая rs1191551, проявляет активность УРЭ, причем один из аллелей rs1191551 активирует транскрипцию значимо лучше другого.
Затем с использованием технологии CRISPR/Cas9 ученые удалили небольшую область генома, содержащую rs1191551. Удаление этой последовательности приводило к ощутимому снижению транскрипции гена FOXG1. Это подтвердило, что rs1191551 находится внутри УРЭ, контролирующего экспрессию гена FOXG1. Таким образом, есть все основания полагать, что один из вариантов rs1191551 приводит к уменьшению экспрессии гена FOXG1 в эмбриональном мозге, что приводит к увеличению предрасположенности носителей этого варианта к шизофрении.
Исследователи из Калифорнии наглядно продемонстрировали, как изучение пространственной укладки ДНК в релевантных клетках может служить основой для расшифровки результатов генетических исследований сложных наследственных заболеваний, в частности шизофрении.
Наложение данных Hi-C на результаты GWAS дает лишь ориентировочный список генов-кандидатов и соответствующих им каузальных полиморфизмов. Нет никаких сомнений в том, что многие среди найденных генов-кандидатов пространственно сближены с регионами GWAS по случайности. Только последующие функциональные исследования помогут подтвердить роль некоторых из обнаруженных генов в развитии шизофрении.
Тем не менее огромная роль подобных работ заключается в сужении потенциально бесконечного пространства для поиска каузальных полиморфизмов и регулируемых ими генов до в общем-то весьма ограниченного набора. Функциональный анализ уже в самом ближайшем будущем выделит из этого набора гены, дисрегуляция которых играет действительно важную роль в развитии болезни. Этот шорт-лист станет базой для поиска молекулярных каскадов и физиологических процессов, изменяющихся при шизофрении. Без нахождения этих каскадов и процессов едва ли стоит ожидать значимых успехов в лекарственной терапии болезни.
Аркадий Голов,
лаборатория Клинической генетики, ФГБНУ НЦПЗ