ПОЛИЭНДОКРИНОПАТИИ
АУТОИММУННЫЕ ПОЛИГЛАНДУЛЯРНЫЕ СИНДРОМЫ – сочетания аутоиммунных поражений сразу нескольких эндокринных желез формируют два клинических синдрома – АПС I, АПС II
Аутоиммунные полигландулярные синдромы (АПС) - это первичное аутоиммунное поражение двух и более периферических эндокринных желез, приводящее к их недостаточности, часто сочетающееся с различными органоспецифическими неэндокринными заболеваниями аутоиммунного генеза
АПС I – моногенное аутосомно-рецессивное заболевание вследствие мутации гена AIRE, кодирующего белок «аутоиммунный регулятор»
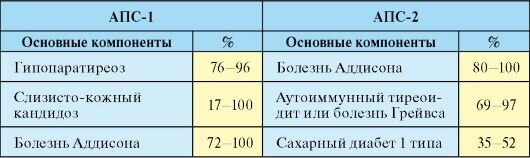
· характерна классическая триада: слизисто-кожный кандидоз, гипопаратиреоз, первичная хроническая надпочечниковая недостаточность (болезнь Аддисона). Для диагноза необходимы только два из этих признаков, а у сибсов – достаточно одного.
Патогенез
В основе патогенеза лежит аутоиммунная деструкция эндокринных желез. При АПС-l с высокой частотой определяются антитела к ферментам надпочечникового стероидогенеза P450scc (20,22-десмолаза), Р450с17 (17α-гидроксилаза) и Р450с21 (21α-гидроксилаза), антитела против панкреатических β-клеток (к глутаматациддекарбоксилазе и L-аминоациддекарбоксилазе) и других пораженных тканей.
Клинические проявления
· 1 признак АПС 1го типа – хронический кандидоз кожи и слизистых (чаще всего до 2х лет) - поражение слизистых оболочек полости рта, гениталий, а также кожи, ногтевых валиков, ногтей
· 2 признак – развитие гипопаратиреоза (чаще до 7-9 лет)
· 3 признак – первичная надпочечниковая недостаточность (чаще до 12 лет)
Если у пациента есть родственник 1 порядка с установленным диагнозом АПС 1 типа или характерными его симптомами, достаточно 1 из 3х перечисленных признаков.
Доп. Признаки:
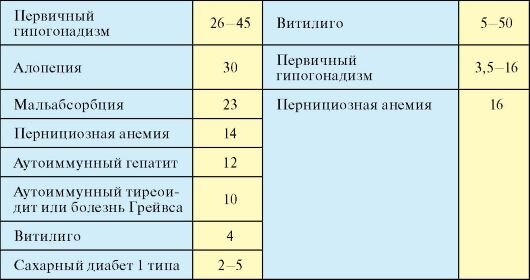
· Алопеция (40%)
· Мальабсорбция (30%)
· У 10-20 % женщин с АПС-l встречается первичный гипогонадизм, развивающийся в результате аутоиммунной деструкции яичников (аутоиммунный оофорит); клинически он проявляется первичной или вторичной аменореей.
· АИТ (30%)
· В12-дефицитная анемия (30%)
· СД1 (20%)
· Первичный гипогонадизм (70%)
· Аутоиммунный гепатит (20%)
· Витилиго, седые волосы (25%)
· Кератоконъюктивит, сухой кератит (10%)
· Дисплазия зубной эмали (30%)
· Гипоплазия ногтей (20%)
· Гипоспления (10%)
Новые симптомы заболевания могут присоединяться в течение жизни.
АПС II – многофакторное заболевание с наследственной предрасположенностью. Существует ассоциация между АПС 2 типа и экспрессией некоторых генов главного комплекса гистосовместимости HLA.
АПС-2 обозначаются различные варианты сочетаний аутоиммунной патологии надпочечников (болезнь Аддисона), щитовидной железы (аутоиммунный тиреоидит (АИТ) или болезнь Грейвса) и сахарного диабета 1 типа (СД-1). Наиболее типичными и частыми вариантами АПС-2 являются синдром Шмидта (сочетание первичного гипокортицизма и гипотиреоза в исходе АИТ) и синдром Карпентера (сочетание СД-1 и АИТ).
АПС-2 примерно в 8 раз чаще встречается у женщин, манифестирует в среднем в возрасте между 20 и 50 годами. АПС II наиболее частый синдром полигландулярной недостаточности.
Основные клинические критерии:
· Первичная ХНН
· Хронический АИТ
· СД 1
+/- гипогонадизм, миастения, целиакия, пернициозная анемия, алопеция, серозит, витилиго…
-Целиакия обычно поддается лечению безглютеновой диетой.
-В отсутствии лечения может иметь место гипоCa-мия (не вследствие гипопаратиреоза), остеопения, а иногда и лимфома ЖКТ.
Диагностика:
1. Анамнез:
a. Близкородственные связи
b. Случаи заболеваний эндокринной системы
c. Ранняя инвалидизация и смерть
2. Обследование физикальное
a. Гипогонадизм: измерение роста, массы, оценка полового развития отражает тяжесть течение АПС
b. Кандидоз и алопеция: состояние ногтей, волос, слизистых оболочек
c. Надпочечниковая недостаточность, гипотиреоз: гиперпигментация, витилиго/сухость кожных покровов
d. Гипопаратиреоз: +с-мы Хвостека и Труссо (гипоCa-мия)
3. Лаборатория
a. Гипопаратиреоз: ионизированный кальций, кальций общий, фосфор и ПТГ
b. НН: калий, натрий, кортизол и АКТГ, АРП. Сандартный тест – синактен-депо (пролонгированная форма синтетического аналога АКТГ). Самым ранним симптомом может стать гипогликемия, снижение потребности в инсулине у СД1.
c. Гипотиреоз: повышение ТТГ. Низкий сТ4
d. Половые железы: ЛГ, ФСГ (резко повышены при первичном гипогонадизме), тестостерона и эстрадиола
e. Недостаточность гормона роста: задержка роста > СТГ-стимулированные тесты (исключение СТГ-недостаточности)
f. СД: глюкоза плазмы, HbA1c, ПГТТ
g. Функция печени: гамма-ГТ, АЛТ, АСТ, ЩФ протеинограмма
h. В-12 деф.анемия
i. Гипокальциемия при АПС II мб вследствие целиакии (реже – из-за гипопаратиреоза)
j. Аутоиммунная природа заболевания: АТ к органам-мишеням – к микросомальной тиропероксидазе, к рТТГ в ЩЖ; к GAD, ICA, IAA клеток ПЖ.
4. Молекулярно-генетическая диагностика: определение мутаций гена AIRE. Рекомендовано всем пациентам с хроническим кандидозом кожи и слизистых оболочек, гипопаратиреозом и НН неустановленной этиологии.
5. Инструментальные исследования:
a. УЗИ органов брюшной полости (признаки гепатита и цирроза печени) и ЩЖ
b. ЭГДС (гастрит, эзофагит, грибковые поражения слиз.оболочки верхних отделов ЖКТ)
c. МРТ и КТ головного мозга (признаки гипопитуитаризма, кальцинатов – синдром Фара)
d. Рентген кистей (оценка костного возраста у детей с задержкой роста)
6. Лечение
a. Симптоматическое
b. Одновременное выявление НН + гипотиреоза = в первую очередь ЗГТ глюко- и минералокортикоидами. Однако ЗГТ левотироксином может усугубить явления некомпенсированной НН и привести к развитию адреналового криза. При назначении заместительной терапии глюкокортикоидами следует иметь в виду, что передозировка может способствовать декомпенсации гипопаратиреоза и спровоцировать гипокальциемию.
c. Гипопаратиреоз: витD + кальций.
d. ГКС угнетают абсорбцию кальция в кишечнике > если у пациента НН + гипопаратирез, уровень кальция в крови мб повышен
e. Кандидоз кожи: флуконазол. Нежелательно применять кетоконазол, т.к. он угнетает синтез стероидных гормонов в надпочечниках (общий ингибитор ферментов цитохрома Р450) и половых желез, может спровоцировать адреналовый криз
7. Для пациентов: осведомить о риске рождения больного ребенка, необходимости обследования родственников первого порядка.