Хотя сегодня существует возможность диагностики синдрома до рождения, зачастую патологию обнаруживают уже при родах. Новорожденный с подобной аномалией хромосом имеет типичные и ярко выраженные пороки в развитии, их наличие может помочь в постановке диагноза даже без дополнительных исследований хромосом – генетического анализа (но его проводят в любом случае, без этих данных диагноз не окончательный).
При рождении выявляются:
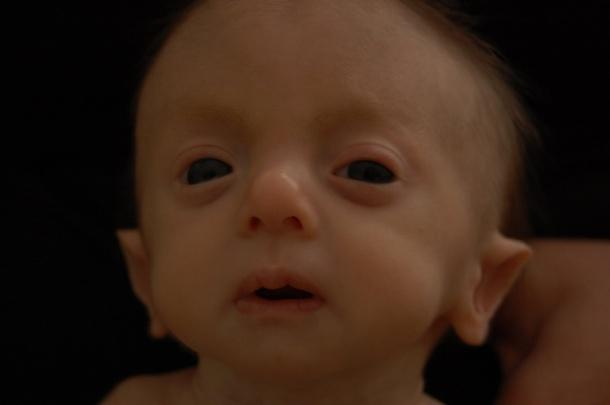
- аномалии в строении формы черепа. Скелет при подобном синдроме страдает наиболее выражено – это проявляется в изменении прежде всего формы черепа с образованием долихоцефалии (резко вытянутый, узкий череп – «башенный»).
Подобная аномалия не типична только для этой патологии, но указывает на имеющиеся отклонения в развитии. Наиболее выражено изменение соотношений по данным измерений в области теменных костей и длины от переносицы до затылочных бугров. Более тяжелой и выраженной аномалией черепа может стать микроцефалия, непропорционально маленький размер мозгового черепа относительно тела.
- специфически измененная форма ушной раковины. Аномальное строение ушных раковин у новорожденных выражено сильно и наблюдаются у 95% малышей, особенно при полном типе синдрома. Расположены ушные раковины гораздо ниже обычного, относительно глазниц они резко опущены, хрящи выпуклые и нет четко сформированных завитков. Мочка и козелок в зачаточном состоянии или полностью отсутствуют, слуховой проход резко сужается и у четверти детей может отсутствовать вовсе.
- аномальное строение неба с нижней челюстью (формирование микрогнатии). У новорожденных, когда подозревают синдром Эдвардса не имеется полного срастания небных отростков в зоне верхней челюсти, что образуют продольную щель. Могут быть варианты с тотальным незаращением только мягкого неба или частичным его сращением, иногда с переходом на губы.

При полной форме порока незаращение может быть двустороннее, верхняя губа расщеплена с двух сторон, дает начало патологическим расщелинам в челюстях и небе, что не дает ребенку полностью закрывать рот. В случае тяжелых патологий возможны сообщения между ротоглоткой и носовой полостями, и в том случае, если рот закрыт. Частота таких аномалий достигает 20-25% и выше.
Могут возникать и другие проблемы – готическое небо (чрезмерно высокий свод костей) и изменения в строении нижней челюсти. Их регистрируют примерно у 70% детей. Типична микрогнатия – сильная втянутость подбородка и нижней челюсти назад, что не дает младенцам полностью закрывать рот и слюна подтекает изо рта. При подобной аномалии возникают трудности с кормлениями, особенно прикладыванием к груди.
- деформация стоп с образованием «стопы-качалки», характерные аномалии длины пальцев конечностей. Изменение стопы типично для 75% и более детей на фоне данного синдрома, возникает изменение положения косточек – таранной, пяточной либо ладьевидной, что приводит к формированию сильно выдающейся пятки на фоне отсутствия свода стопы. Он может быть выгнут вперед, придавая стопам вид ножек от кресла-качалки. Также выявляются и диспропорции в длине пальцев на ногах, особенно большого. Он короче второго пальца на ноге, что видно при распрямленных пальчиках.
- кисти в флексорном положении и сращения между пальцами, особые дерматоглифические рисунки. Синдактилия (этот термин обозначет сращение пальцев) выявляют в 45% случаев, обычно это пальцы на ногах, но могут затрагиваться и руки. При легкой степени кожа создает перепонки, в тяжелых могут образовываться костные перемычки. Также могут быть флексорные аномалии кистей – положение пальцев в аномальном, постоянно напряженном положении, прикрывая мизинцем и большим пальцем все остальные, плотно прижатые к ладошке. Типичны подобные расстройства для 90% детей с синдромом.
Кроме всего прочего, при трисомии типичны особые узоры на пальцах и ладошках с особой формой кожных складок. При развит этого синдрома отклонения типичны для 60% малышей. Это очень частые дуги на подушечках, отсутствие кожной складки между крайней и предпоследней фалангами, развитие поперечный бороздки – она имеет термин «обезьянья» линия.
- пороки гениталий. У мальчиков это недоразвитие пениса и мошонки, у девочек увеличение размеров клитора. Аномалии сочетаются обычно с пороками мочеполовой системы, но внешне остальные признаки не увидеть.
Помимо этих проявлений есть и более мелкие признаки и аномалии развития детей, которые могут в предварительном диагностировании синдрома. Важно отметить, что один из перечисленных признаков и даже два еще не говорят о диагнозе, при синдроме пороки множественные и имеют различные сочетания. Большинство их описанных проявлений могут встречаться как единичные варианты и диагноза не подтверждают, они могут быть изолированным вариантом отклонений в течении беременности или проявлением иных патологий. Диагноз только по внешним порокам ставить нельзя!!!
Более того, на фоне неполного или мозаичного типов синдрома внешних признаков может быть немного – один-два, при этом есть серьезные пороки внутренних органов с частыми отклонениями в хромосомах. Именно они и определяют дальнейшие прогнозы на жизнь и течение синдрома.
Развитие детей: особенности синдрома
Только внешним видом при появлении на свет и проявлениями пороков в развития, аномалия Эдвардса не ограничивается, по мере роста и развития возможны проявления все новых и более тяжелых отклонений. Первые из них можно выявить через несколько недель с момента рождения.
Нередко по ним впервые можно заподозрить диагноз при мозаичном и неполном типе патологии, если внешних дефектов мало или практически нет. Описанные выше пороки и внешние изменения становятся по мере роста более резкими и выраженными, постепенно добавляются новые проблемы и аномалии, которые при рождении не были заметны.
Так, сюда стоит отнести:
- Резкое и выраженное отставание в уровне физического развития. При рождении масса таких детей не превышает 2-2.5 кг, и причиной тому являются хромосомные дефекты, которые не дают полноценно развиваться системам и органам. По мере развития показатели роста и массы тела по месяцам существенно отличаются от норм, также слабо прибывают и окружности груди. Размеры головы могут прибывать но норме или выше нее, что может указывать на скопление внутри полости черепа избытка жидкости, формируя гидроцефалию.
- Проблемы со стопами. По мере роста формируется косолапость в силу аномалий строения костей и связок, мышечного гипертонуса. Страдает и контроль за развитием стопы и тонусом от нервной системы. Это угрожает тем, что дети поздно начинают ходить или не могут этого делать вовобще в силу общего тяжелого состояния. Внешне косолапость проявляется в выраженной деформации стопы и ненормальной ее установке в покое.
- Нарушение мышечного тонуса. При рождении гипертонус типичен для кистей, формируя их флексорное положение, но по мере развития распространяется на соседние групп мышц, приводя к вычурным позам. Но чаще тонус мышц снижен, дети вялые и атоничные, конечности весят плетьми. Могут также быть состояния дистонии с резким сокращением отдельных мышечных групп на фоне гипотонии других. При этом руки могут быть согнуты, а ноги – переразгибаются. Это приводит к нарушению координации и хаотичности движений, вывихам и ненормальному положению конечностей.
- Атипичные реакции нервной системы, проблемы эмоциональности. Некоторые отделы мозга у детей с синдромом Эдвардса недоразвиты, в связи с чем формируются проблемы эмоциональности и расторможенности, неадекватные реакции на окружающее. Это связано обычно с мозолистым телом и мозжечковыми гипоплазиями, что формирует также отставание умственного развития. Внешне это проявляется в отсутствующем взоре, неэмоциональности и невозможности установления контакта, отсутствии слежения за объектами. Дети не реагируют на звуки в силу проблем с ушами и нервной системой, подобные аномалии выявляют в первые же месяцы жизни.
В целом, в силу тяжелых и комбинированных поражений дети не доживают до периода совершеннолетия. Часть их при мозаичной форме может достигать возраста подростков с глубокими степенями имбецильности и внешними проявлениями пороков.
Методы диагностики синдрома
На сегодняшний день существует три типа диагностик синдрома, которые принципиально отличаются друг от друга. Учитывая тот факт, что патология неизлечима и летальна в раннем возрасте, важно обращать внимание на пренатальную диагностику, которая не допускает рождения детей с данными аномалиями. Полезными будут и консультации врачей-генетиков в семьях, где имели место случаи хромосомных патологий. Можно провести:
- Диагностику половых клеток еще до зачатия. К сожалению, подобный метод применим еще очень ограниченно и только в протоколах ЭКО при предварительном изучении половых клеток. если же это естественное зачатие, врачи могут прогнозировать более высокие риски рождения таких детей, но это не более, чем прогнозы. Точно узнать – родится ли такой ребенок или нет – невозможно. Исследуется семейный анамнез с учетом всех имеющихся заболеваний, а также оцениваются факторы риска, а также проводится генетический анализ обоих родителей с применением сложной аппаратуры и анализов. За счет определения кариотипа родителей могут делаться определенные выводы.
- Выявление синдрома в период гестации. При развитии плода существует несколько методов прямого или косвенного подтверждения синдрома Эдвардса, изначально для этих целей применяют УЗИ-скрининг и анализы крови (включая НИПТ) с изучением особых показателей, а при высоких рисках проводятся уже инвазивные процедуры – амниоцентез, либо кордоцентез и биопсия ворсинок хориона.
Проводят скрининги в строго определенные сроки, когда изменение данных УЗИ и показателей крови будет наиболее значимым. Получение непосредственно клеток плода или его крови позволяет провести исследование его хромосом и подтвердить или опровергнуть у него синдром Эдвардса. Это помогает в принятии решения о пролонгации или прерывании беременности по медицинским показаниям.
- Подтверждение синдрома после рождения. Его проводят по результатам внешних данных в комбинации с рядом генетических анализов, подтверждающих наличие трисомии по 18-ой паре хромосом. Помимо этого важно оценить состояние ребенка и дать прогнозы в отношении его жизни и состояния здоровья, чтобы понять, какой объем помощи ему требуется для выживания. Без медицинской помощи при подобном синдроме дети могут быстро погибать поле рождения в ближайшие недели. Для выявления пороков во внутренних органах проводят УЗИ и ЭХО-КГ, рентгенографию и ЭЭГ, целый ряд анализов крови и мочи, дополнительные исследования вплоть до МРТ и КТ.
Лечение и прогнозы
К сожалению, данный синдром на сегодняшний день неизлечим, и врачи могут помочь ребенку только лучше справляться с различными проблемами за счет оперативных вмешательств и консервативной терапии. Обычно дети доживают до нескольких месяцев или года, только при частичной или мозаичной форме могут прожить более длительно. Гибель наступает от множественных пороков развития и аномалий в работе органов, а также присоединения сопутствующих осложнений. радикально исправить дефекты хромосом в клетках тела ребенка на сегодняшнем этапе развития медицины невозможно.